

Description
48,XXXY syndrome is a chromosomal condition in boys and men that causes intellectual disability, developmental delays, physical differences, and an inability to father biological children (infertility). Its signs and symptoms vary among affected individuals.
Most boys and men with 48,XXXY syndrome have mild intellectual disability with learning difficulties. Speech and language development is particularly affected. Most affected boys and men can understand what other people say more easily than they themselves can speak. The problems with speech and communication can contribute to behavioral issues, including irritability and outbursts or temper tantrums. Boys and men with 48,XXXY syndrome tend to have anxiety, a short attention span, and impaired social skills.
48,XXXY syndrome is also associated with weak muscle tone (hypotonia) and problems with coordination that delay the development of motor skills, such as sitting, standing, and walking. Affected boys and men tend to be taller than their peers, with an average adult height of over 6 feet.
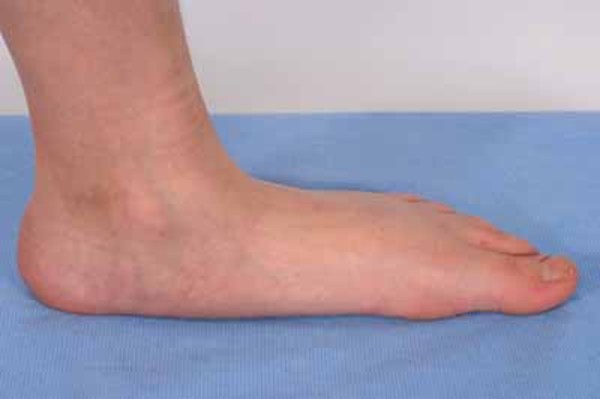
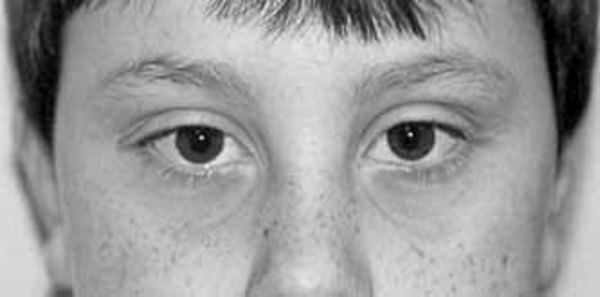
Other physical differences associated with 48,XXXY syndrome include abnormal fusion of certain bones in the forearm (radioulnar synostosis), an unusually large range of joint movement (hyperextensibility), elbow abnormalities, curved pinky fingers (fifth finger clinodactyly), and flat feet (pes planus). Affected individuals may have distinctive facial features, including widely spaced eyes (ocular hypertelorism), outside corners of the eyes that point upward (upslanting palpebral fissures), and skin folds covering the inner corner of the eyes (epicanthal folds). However, some boys and men with 48,XXXY syndrome do not have these differences in their facial features.

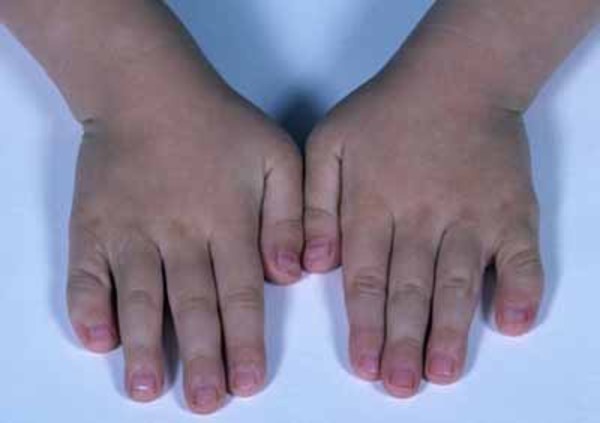

48,XXXY syndrome disrupts male sexual development. The penis is shorter than usual, and the testes may be undescended, which means they are abnormally located inside the pelvis or abdomen. The testes are small and do not produce enough testosterone, which is the hormone that directs male sexual development. The shortage of testosterone often leads to incomplete puberty. Starting in adolescence, affected boys and men may have sparse body hair, and some experience breast enlargement (gynecomastia). Their testes typically do not produce sperm, so most men with this condition are infertile.
Frequency
48,XXXY syndrome affects between 1 in 17,000 and 1 in 50,000 newborn boys. It is a relatively uncommon sex chromosome disorder, which is a group of conditions caused by changes in the number of sex chromosomes (the X chromosome and the Y chromosome).

Causes
48,XXXY syndrome is a sex chromosome disorder in boys and men that results from having two extra X chromosomes in each cell. People typically have 46 chromosomes in each cell, two of which are the sex chromosomes. Females have two X chromosomes (46,XX), and males have one X chromosome and one Y chromosome (46,XY). Boys and men with 48,XXXY syndrome have the usual single Y chromosome, but they have three copies of the X chromosome, for a total of 48 chromosomes in each cell.
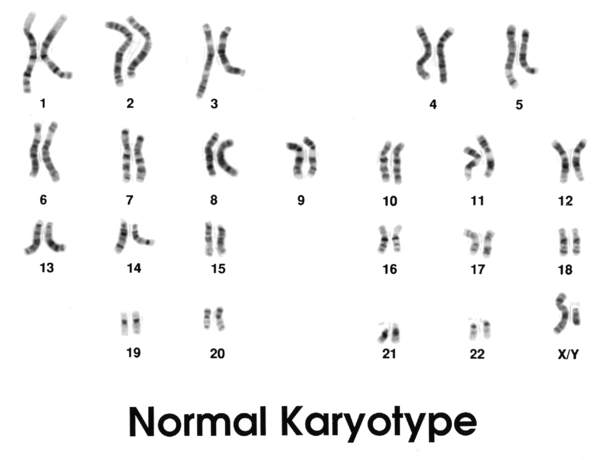
Boys and men with 48,XXXY syndrome have extra copies of multiple genes on the X chromosome. The activity of these extra genes affects many aspects of development, including sexual development before birth and at puberty. Researchers are working to determine which genes contribute to the specific developmental and physical differences that occur with 48,XXXY syndrome.
48,XXXY syndrome is sometimes described as a variant of another sex chromosome disorder called Klinefelter syndrome. Boys and men with Klinefelter syndrome have one extra copy of the X chromosome, for a total of 47 chromosomes in each cell (47,XXY). Like 48,XXXY syndrome, Klinefelter syndrome affects male sexual development and can be associated with learning disabilities and problems with speech and language development. However, the features of 48,XXXY syndrome tend to be more severe than those of Klinefelter syndrome and affect more parts of the body. As doctors and researchers have learned more about the differences between these sex chromosome disorders, they have started to consider them as separate conditions.
Inheritance
This condition is not inherited; it occurs as a random event during the formation of reproductive cells (eggs or sperm) in one of an affected person's parents. During cell division, an error called nondisjunction prevents X chromosomes from being distributed normally among reproductive cells as they form. Typically, as cells divide, each egg cell gets a single X chromosome, and each sperm cell gets either an X chromosome or a Y chromosome. However, because of nondisjunction, an egg cell or a sperm cell can also end up with two extra copies of the X chromosome.
If an egg cell with two extra X chromosomes (XXX) is fertilized by a sperm cell with one Y chromosome, the resulting child will have 48,XXXY syndrome. Similarly, if a sperm cell with a Y chromosome and two extra X chromosomes (XXY) fertilizes an egg cell with a single X chromosome, the resulting child will have 48,XXXY syndrome.
Other Names for This Condition
- XXXY males
- XXXY syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Scientific Articles on PubMed
References
- Gropman A, Samango-Sprouse CA. Neurocognitive variance and neurological underpinnings of the X and Y chromosomal variations. Am J Med Genet C Semin Med Genet. 2013 Feb 15;163C(1):35-43. doi: 10.1002/ajmg.c.31352. Epub 2013 Jan 18. Citation on PubMed
- Tartaglia N, Ayari N, Howell S, D'Epagnier C, Zeitler P. 48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr. 2011 Jun;100(6):851-60. doi: 10.1111/j.1651-2227.2011.02235.x. Epub 2011 Apr 8. Citation on PubMed or Free article on PubMed Central
- Visootsak J, Graham JM Jr. Social function in multiple X and Y chromosome disorders: XXY, XYY, XXYY, XXXY. Dev Disabil Res Rev. 2009;15(4):328-32. doi: 10.1002/ddrr.76. Citation on PubMed or Free article on PubMed Central
- Visootsak J, Rosner B, Dykens E, Tartaglia N, Graham JM Jr. Behavioral phenotype of sex chromosome aneuploidies: 48,XXYY, 48,XXXY, and 49,XXXXY. Am J Med Genet A. 2007 Jun 1;143A(11):1198-203. doi: 10.1002/ajmg.a.31746. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.