

Description
2q37 deletion syndrome is a condition that can affect many parts of the body. Most babies with 2q37 deletion syndrome are born with weak muscle tone (hypotonia), which usually improves with age. Other neurological abnormalities that are common in affected individuals include mild to severe intellectual disability; delayed development of motor skills, such as sitting and walking; and behavioral problems. About 25 percent of people with this condition have autism spectrum disorder, a developmental condition that affects communication and social interaction.
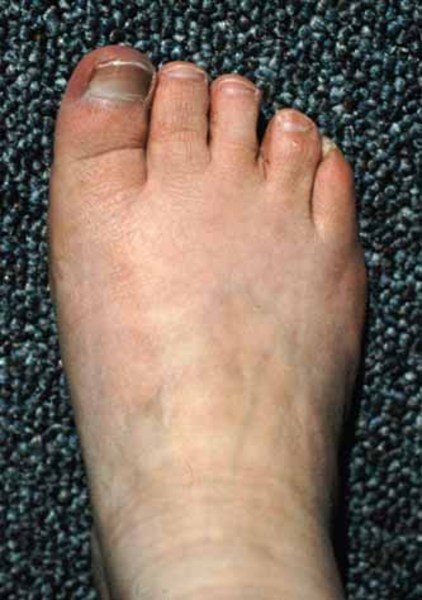
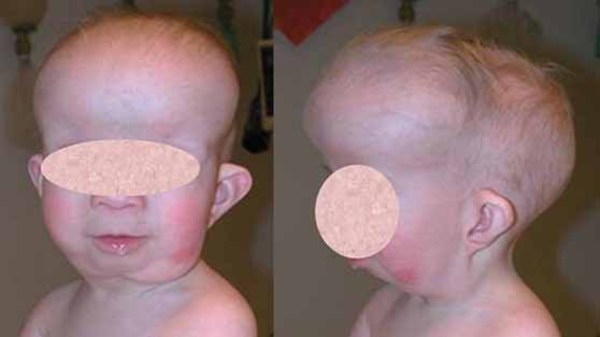
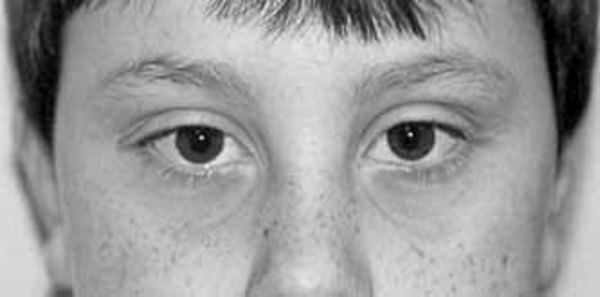
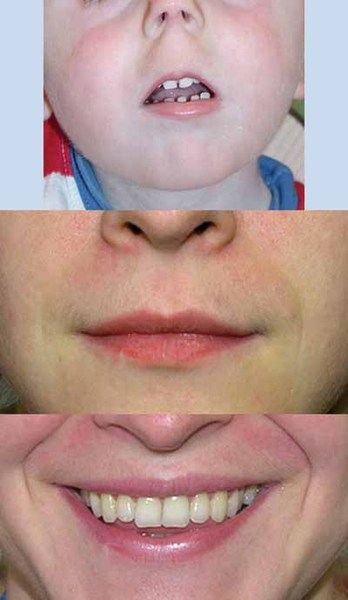
Unusual physical features are also common in people with 2q37 deletion syndrome. About half of affected individuals have unusually short fingers and toes (brachydactyly), often with abnormally short fourth toes that may overlap the other toes. Additional features of this condition may include short stature, obesity, or sparse hair. Many people with 2q37 deletion syndrome have characteristic facial features that can include a prominent forehead, a low frontal hairline, thin eyelids, skin folds covering the inner corner of the eyes (epicanthal folds), outside corners of the eyes that point upward (upslanting palpebral fissures), a small nose, a small mouth with thin lips, a smooth space between the upper lip and nose (smooth philtrum), prominent cheekbones, a large chin, and minor ear abnormalities.


Other features of 2q37 deletion syndrome can include seizures and an inflammatory skin disorder called eczema. Some affected individuals have malformations of the brain, heart, gastrointestinal system, kidneys, or genitalia. A few people with 2q37 deletion syndrome develop a rare form of kidney cancer called Wilms tumor.
Frequency
2q37 deletion syndrome appears to be a rare condition, although its exact prevalence is unknown. At least 115 cases have been reported worldwide.
Causes
2q37 deletion syndrome is caused by deletions of genetic material from a specific region in the long (q) arm of chromosome 2. The deletions occur near the end of the chromosome at a location designated 2q37. The size of the deletion varies among affected individuals, with most affected people missing 2 million to 9 million DNA building blocks (also written as 2 Mb to 9 Mb).
Researchers are working to identify all of the genes whose loss contributes to the features of 2q37 deletion syndrome. Many of these genes have not been well characterized. However, genes in this region appear to be critical for the normal development of many parts of the body.
Researchers have determined that loss of a particular gene on chromosome 2, called HDAC4, is likely to account for many of the syndrome's characteristic signs (such as intellectual disability and skeletal abnormalities). While the deleted segment in 2q37 deletion syndrome varies in size, it always contains the HDAC4 gene. Additionally, a few people with mutations in only the HDAC4 gene have many of the features of 2q37 deletion syndrome. It is unclear what role the other genes on 2q37 play in this disorder.
Inheritance
Most cases of 2q37 deletion syndrome are not inherited. They result from a chromosomal deletion that occurs as a random event during the formation of reproductive cells (eggs or sperm) or in early fetal development. Affected people typically have no history of the disorder in their family.
Rarely, an affected individual inherits a copy of chromosome 2 with a deleted segment from an affected parent. In these cases, the parent is usually less severely affected than the child, for reasons that are unknown. When an affected child inherits a chromosomal deletion from a parent, it is inherited in an autosomal dominant pattern, which means one copy of the altered chromosome in each cell is sufficient to cause the disorder.

Other Names for This Condition
- 2q37 microdeletion syndrome
- Albright hereditary osteodystrophy-like syndrome
- Brachydactyly-mental retardation syndrome
- Chromosome 2q37 deletion syndrome (disorder)
- Deletion 2q37
- Monosomy 2q37
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aldred MA, Sanford RO, Thomas NS, Barrow MA, Wilson LC, Brueton LA, Bonaglia MC, Hennekam RC, Eng C, Dennis NR, Trembath RC. Molecular analysis of 20 patients with 2q37.3 monosomy: definition of minimum deletion intervals for key phenotypes. J Med Genet. 2004 Jun;41(6):433-9. doi: 10.1136/jmg.2003.017202. No abstract available. Citation on PubMed or Free article on PubMed Central
- Casas KA, Mononen TK, Mikail CN, Hassed SJ, Li S, Mulvihill JJ, Lin HJ, Falk RE. Chromosome 2q terminal deletion: report of 6 new patients and review of phenotype-breakpoint correlations in 66 individuals. Am J Med Genet A. 2004 Nov 1;130A(4):331-9. doi: 10.1002/ajmg.a.30156. Citation on PubMed
- Fisch GS, Falk RE, Carey JC, Imitola J, Sederberg M, Caravalho KS, South S. Deletion 2q37 syndrome: Cognitive-behavioral trajectories and autistic features related to breakpoint and deletion size. Am J Med Genet A. 2016 Sep;170(9):2282-91. doi: 10.1002/ajmg.a.37782. Epub 2016 Jun 9. Citation on PubMed
- Jean-Marcais N, Decamp M, Gerard M, Ribault V, Andrieux J, Kottler ML, Plessis G. The first familial case of inherited 2q37.3 interstitial deletion with isolated skeletal abnormalities including brachydactyly type E and short stature. Am J Med Genet A. 2015 Jan;167A(1):185-9. doi: 10.1002/ajmg.a.36428. Epub 2014 Nov 17. Citation on PubMed
- Leroy C, Landais E, Briault S, David A, Tassy O, Gruchy N, Delobel B, Gregoire MJ, Leheup B, Taine L, Lacombe D, Delrue MA, Toutain A, Paubel A, Mugneret F, Thauvin-Robinet C, Arpin S, Le Caignec C, Jonveaux P, Beri M, Leporrier N, Motte J, Fiquet C, Brichet O, Mozelle-Nivoix M, Sabouraud P, Golovkine N, Bednarek N, Gaillard D, Doco-Fenzy M. The 2q37-deletion syndrome: an update of the clinical spectrum including overweight, brachydactyly and behavioural features in 14 new patients. Eur J Hum Genet. 2013 Jun;21(6):602-12. doi: 10.1038/ejhg.2012.230. Epub 2012 Oct 17. Citation on PubMed or Free article on PubMed Central
- Morris B, Etoubleau C, Bourthoumieu S, Reynaud-Perrine S, Laroche C, Lebbar A, Yardin C, Elsea SH. Dose dependent expression of HDAC4 causes variable expressivity in a novel inherited case of brachydactyly mental retardation syndrome. Am J Med Genet A. 2012 Aug;158A(8):2015-20. doi: 10.1002/ajmg.a.35463. Epub 2012 Jun 29. Citation on PubMed
- Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010 Aug 13;87(2):219-28. doi: 10.1016/j.ajhg.2010.07.011. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.