

Normal Function
The RHO gene provides instructions for making a protein called rhodopsin. This protein is found in the light-sensitive tissue at the back of the eye (the retina). The retina contains two types of specialized light receptor cells called rods and cones. Cones are responsible for color vision and vision in bright light. Rods are responsible for vision in low light. Rhodopsin is found in rods, where it plays a critical role in vision in low-light conditions.
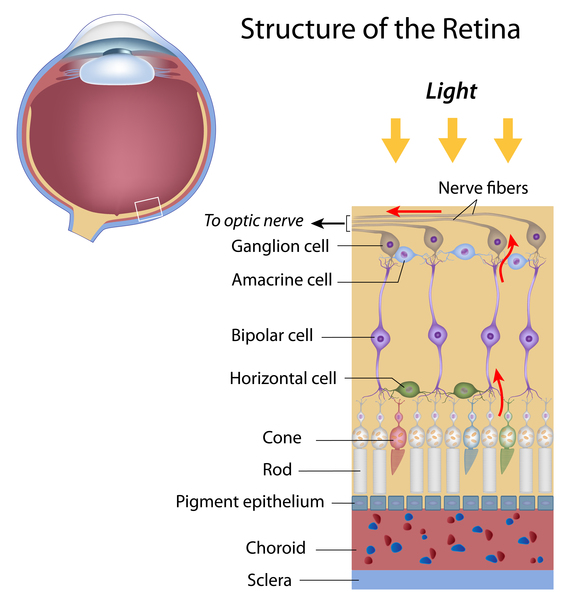
The rhodopsin protein binds to a molecule called 11-cis retinal, which is a form of vitamin A. When light hits this molecule, it activates rhodopsin and sets off a series of chemical reactions that create electrical signals. These signals are transmitted to the brain for interpretation.
Health Conditions Related to Genetic Changes
Autosomal dominant congenital stationary night blindness
Genetic changes that cause disease are called pathogenic variants. Pathogenic variants in the RHO gene can cause autosomal dominant congenital stationary night blindness, which is characterized by a loss of vision in low light that remains stable (stationary) over time. Unlike retinitis pigmentosa (described below), autosomal dominant congenital stationary night blindness does not affect daytime vision. Autosomal dominant means that a pathogenic variant in one copy of the RHO gene in each cell is sufficient to cause the disorder.
The RHO gene variants that are associated with autosomal dominant congenital stationary night blindness cause cells to make a version of rhodopsin that can activate even when no light is present. Because rhodopsin no longer needs light to activate, rod cell signaling is disrupted and the signals sent to the brain are misinterpreted. This makes it difficult for affected individuals to see in low-light conditions.
More About This Health ConditionRetinitis pigmentosa
Pathogenic variants in the RHO gene can also cause retinitis pigmentosa, which is characterized by vision loss that worsens over time. Retinitis pigmentosa affects cells in the retina. RHO gene variants are a common cause of cases of retinitis pigmentosa that are inherited in an autosomal dominant manner. In rare cases, variants in the RHO gene cause autosomal recessive retinitis pigmentosa. Autosomal recessive means that variants in both copies of the RHO gene in each cell are needed to cause the condition.
Most of the RHO gene variants that are responsible for retinitis pigmentosa lead to an altered version of rhodopsin that interferes with essential cell functions, causing rods to self-destruct. Because rods are essential for vision under low-light conditions, the loss of these cells leads to progressive night blindness in people with retinitis pigmentosa.
Retinitis pigmentosa is also associated with a gradual loss of cone cells. Researchers are working to learn why variants in the RHO gene that lead to the destruction of rod cells also lead to the destruction of cone cells in people with retinitis pigmentosa.
More About This Health ConditionOther disorders
Pathogenic variants in the RHO gene can also cause retinitis punctata albescens, a condition that is characterized by night blindness that begins in childhood. Some researchers consider retinitis punctata albescens to be a form of retinitis pigmentosa. Individuals with retinitis punctata albescens typically have white dots, also called flecks, that can be seen in the retina during an eye exam. The dots are present in childhood and indicate a breakdown of cells. The variants in the RHO gene that are associated with retinitis punctata albescens cause cells to make an altered version of rhodopsin, which impairs the function of rod cells. This causes the night blindness that is seen in the early stages of the condition; eventually, the function of neighboring cells becomes impaired, which can lead to vision loss.
Other Names for This Gene
- CSNBAD1
- OPN2
- opsin
- RP4
Additional Information & Resources
Tests Listed in the Genetic Testing Registry
Scientific Articles on PubMed
Catalog of Genes and Diseases from OMIM
References
- Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990 Jan 25;343(6256):364-6. doi: 10.1038/343364a0. Citation on PubMed
- Fahim AT, Daiger SP, Weleber RG. Nonsyndromic Retinitis Pigmentosa Overview. 2000 Aug 4 [updated 2023 Apr 6]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1417/ Citation on PubMed
- Fain GL. The mechanism of genetically inherited night blindness. Proc Natl Acad Sci U S A. 2024 May 28;121(22):e2408254121. doi: 10.1073/pnas.2408254121. Epub 2024 May 20. No abstract available. Citation on PubMed
- Foote KG, Wong JJ, Boehm AE, Bensinger E, Porco TC, Roorda A, Duncan JL. Comparing Cone Structure and Function in RHO- and RPGR-Associated Retinitis Pigmentosa. Invest Ophthalmol Vis Sci. 2020 Apr 9;61(4):42. doi: 10.1167/iovs.61.4.42. Citation on PubMed
- McAlear SD, Kraft TW, Gross AK. 1 rhodopsin mutations in congenital night blindness. Adv Exp Med Biol. 2010;664:263-72. doi: 10.1007/978-1-4419-1399-9_30. Citation on PubMed
- Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005 Apr;11(4):177-85. doi: 10.1016/j.molmed.2005.02.007. Citation on PubMed
- Morris MB, Dastmalchi S, Church WB. Rhodopsin: structure, signal transduction and oligomerisation. Int J Biochem Cell Biol. 2009 Apr;41(4):721-4. doi: 10.1016/j.biocel.2008.04.025. Epub 2008 Aug 3. Citation on PubMed
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000 Aug 4;289(5480):739-45. doi: 10.1126/science.289.5480.739. Citation on PubMed
- Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743-67. doi: 10.1146/annurev.biochem.75.103004.142743. Citation on PubMed or Free article on PubMed Central
- Ripps H. Cell death in retinitis pigmentosa: gap junctions and the 'bystander' effect. Exp Eye Res. 2002 Mar;74(3):327-36. doi: 10.1006/exer.2002.1155. Citation on PubMed
- Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002 Jul 15;115(Pt 14):2907-18. doi: 10.1242/jcs.115.14.2907. Citation on PubMed
- Smith SO. Structure and activation of the visual pigment rhodopsin. Annu Rev Biophys. 2010;39:309-28. doi: 10.1146/annurev-biophys-101209-104901. Citation on PubMed
- Souied E, Soubrane G, Benlian P, Coscas GJ, Gerber S, Munnich A, Kaplan J. Retinitis punctata albescens associated with the Arg135Trp mutation in the rhodopsin gene. Am J Ophthalmol. 1996 Jan;121(1):19-25. doi: 10.1016/s0002-9394(14)70530-6. Citation on PubMed
- Zeitz C, Gross AK, Leifert D, Kloeckener-Gruissem B, McAlear SD, Lemke J, Neidhardt J, Berger W. Identification and functional characterization of a novel rhodopsin mutation associated with autosomal dominant CSNB. Invest Ophthalmol Vis Sci. 2008 Sep;49(9):4105-14. doi: 10.1167/iovs.08-1717. Epub 2008 May 16. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.