

Description
Woodhouse-Sakati syndrome is a disorder that primarily affects the body's network of hormone-producing glands (the endocrine system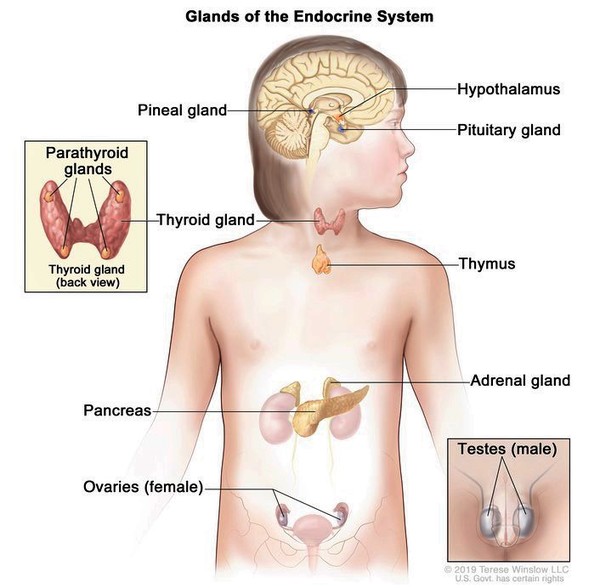 ) and the nervous system. The signs and symptoms of this condition vary widely among affected individuals, even within the same family.
) and the nervous system. The signs and symptoms of this condition vary widely among affected individuals, even within the same family.
People with Woodhouse-Sakati syndrome produce abnormally low amounts of hormones that direct sexual development (hypogonadism), which typically becomes apparent during adolescence. Without hormone replacement therapy, affected individuals do not develop secondary sexual characteristics such as pubic hair, breast growth in women, or a deepening voice in men. Women with Woodhouse-Sakati syndrome do not have functional ovaries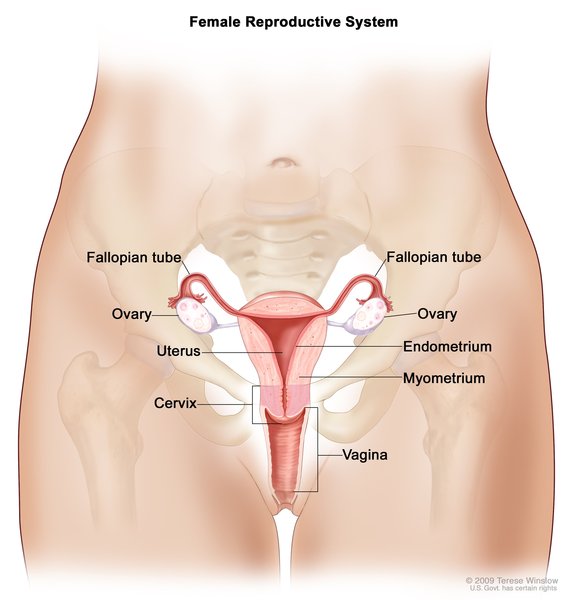 and may instead have undeveloped tissues called streak gonads. The uterus may also be small or absent. Men with this disorder have testes
and may instead have undeveloped tissues called streak gonads. The uterus may also be small or absent. Men with this disorder have testes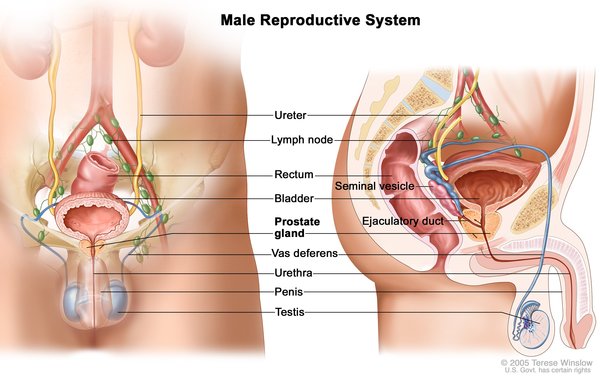 that produce little to no sperm
that produce little to no sperm . As a result, people with Woodhouse-Sakati syndrome have difficulty having biological children (a condition called infertility).
. As a result, people with Woodhouse-Sakati syndrome have difficulty having biological children (a condition called infertility).
Some affected individuals have certain characteristic facial features, including a long, triangular face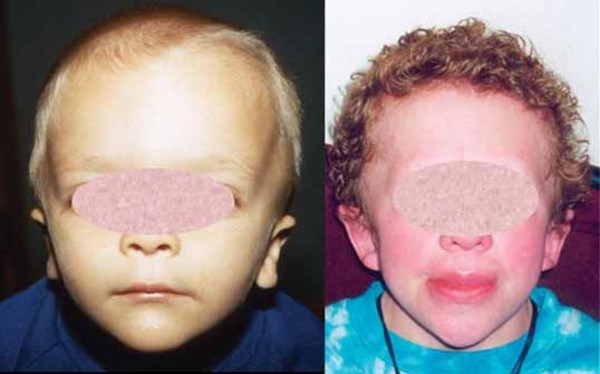 ; widely spaced eyes (hypertelorism
; widely spaced eyes (hypertelorism ); and a prominent bridge of the nose. People with Woodhouse-Sakati syndrome also experience hair loss (alopecia) that begins in childhood and worsens over time. Eyelashes and eyebrows are sparse or absent, and affected men have little or no facial hair. By their mid-twenties, almost all affected individuals develop diabetes mellitus, and they may also have reduced production of thyroid
); and a prominent bridge of the nose. People with Woodhouse-Sakati syndrome also experience hair loss (alopecia) that begins in childhood and worsens over time. Eyelashes and eyebrows are sparse or absent, and affected men have little or no facial hair. By their mid-twenties, almost all affected individuals develop diabetes mellitus, and they may also have reduced production of thyroid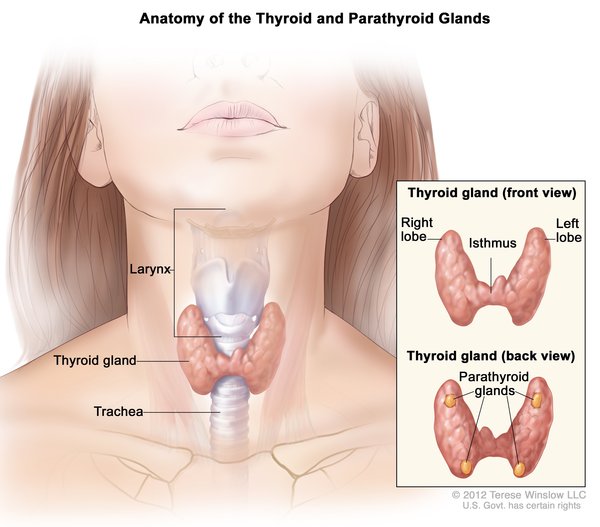 hormones (hypothyroidism).
hormones (hypothyroidism).
Individuals with Woodhouse-Sakati syndrome may have neurological problems. A group of movement abnormalities called dystonias are common in affected individuals, and they generally begin in adolescence or young adulthood. These movement abnormalities can include involuntary tensing of the muscles (muscle contractions) or twisting of specific body parts such as an arm or a leg. Other neurological features can include difficulty with speech (dysarthria) or swallowing (dysphagia), and mild intellectual disabilities.
Changes in the inner ears can lead to hearing loss (sensorineural hearing loss) in people with Woodhouse-Sakati syndrome. The hearing loss can range from mild to total. This loss usually occurs in adolescence.
can lead to hearing loss (sensorineural hearing loss) in people with Woodhouse-Sakati syndrome. The hearing loss can range from mild to total. This loss usually occurs in adolescence.
In some affected individuals, abnormal deposits of iron in the brain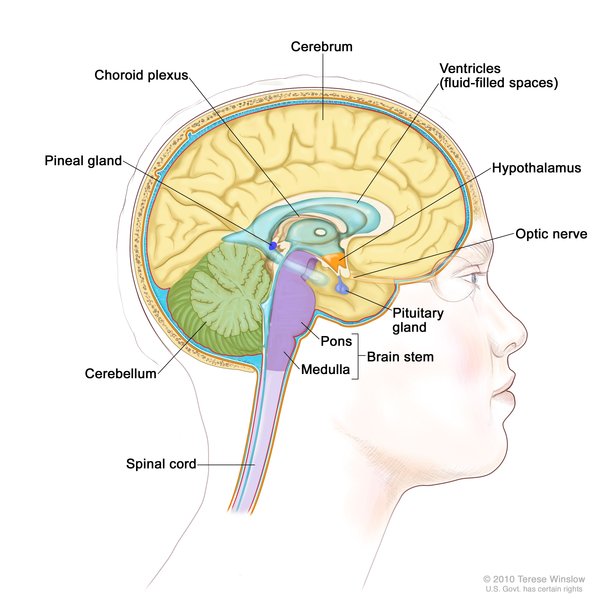 have been detected with medical imaging. For this reason, Woodhouse-Sakati syndrome is sometimes classified as part of a group of disorders called neurodegeneration with brain iron accumulation (NBIA).
have been detected with medical imaging. For this reason, Woodhouse-Sakati syndrome is sometimes classified as part of a group of disorders called neurodegeneration with brain iron accumulation (NBIA).
Some researchers classify Woodhouse-Sakati syndrome into two types, depending on the signs and symptoms. People with Woodhouse-Sakati syndrome type 1 tend to have more severe neurological problems, and those with type 2 have milder or no neurological problems.
Frequency
Woodhouse-Sakati syndrome is a rare disorder. More than 180 affected individuals, largely from Greater Middle East countries, have been described in the medical literature.
Causes
Woodhouse-Sakati syndrome is caused by variants (also called mutations) in the DCAF17 gene. This gene provides instructions for making a protein whose function is unknown. The protein is found in several organs and tissues in the body, including the brain, skin, and liver .
.
Most of the DCAF17 gene variants that have been identified in people with Woodhouse-Sakati syndrome result in a protein that is abnormally short and breaks down quickly or whose usual function is impaired. The loss of DCAF17 protein function likely accounts for the features of Woodhouse-Sakati syndrome, although it is unclear how a shortage of this protein leads to hormone abnormalities and the other signs and symptoms. Researchers suggest that the different features of the disorder may be caused by variations in other genes called modifiers; however, these genes have not been identified.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Diabetes-hypogonadism-deafness-intellectual disability syndrome
- WSS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Abusrair AH, Bohlega S, Al-Semari A, Al-Ajlan FS, Al-Ahmadi K, Mohamed B, AlDakheel A. Brain MR Imaging Findings in Woodhouse-Sakati Syndrome. AJNR Am J Neuroradiol. 2018 Dec;39(12):2256-2262. doi: 10.3174/ajnr.A5879. Epub 2018 Nov 8. Citation on PubMed
- Agopiantz M, Corbonnois P, Sorlin A, Bonnet C, Klein M, Hubert N, Pascal-Vigneron V, Jonveaux P, Cuny T, Leheup B, Weryha G. Endocrine disorders in Woodhouse-Sakati syndrome: a systematic review of the literature. J Endocrinol Invest. 2014 Jan;37(1):1-7. doi: 10.1007/s40618-013-0001-5. Epub 2014 Jan 8. Citation on PubMed
- Alazami AM, Al-Saif A, Al-Semari A, Bohlega S, Zlitni S, Alzahrani F, Bavi P, Kaya N, Colak D, Khalak H, Baltus A, Peterlin B, Danda S, Bhatia KP, Schneider SA, Sakati N, Walsh CA, Al-Mohanna F, Meyer B, Alkuraya FS. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am J Hum Genet. 2008 Dec;83(6):684-91. doi: 10.1016/j.ajhg.2008.10.018. Epub 2008 Nov 20. Citation on PubMed or Free article on PubMed Central
- Alazami AM, Schneider SA, Bonneau D, Pasquier L, Carecchio M, Kojovic M, Steindl K, de Kerdanet M, Nezarati MM, Bhatia KP, Degos B, Goh E, Alkuraya FS. C2orf37 mutational spectrum in Woodhouse-Sakati syndrome patients. Clin Genet. 2010 Dec;78(6):585-90. doi: 10.1111/j.1399-0004.2010.01441.x. Citation on PubMed
- Bohlega S, Abusrair AH, Al-Ajlan FS, Alharbi N, Al-Semari A, Bohlega B, Abualsaud D, Alkuraya F. Patterns of neurological manifestations in Woodhouse-Sakati Syndrome. Parkinsonism Relat Disord. 2019 Dec;69:99-103. doi: 10.1016/j.parkreldis.2019.10.007. Epub 2019 Oct 13. Citation on PubMed
- Bohlega SA, Abusrair A. Woodhouse-Sakati Syndrome. 2016 Aug 4 [updated 2021 Jul 8]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK378974/ Citation on PubMed
- Chen G, Zhou L, Chen Q, Wang J, Jiang P, Shen R, Long M, Zhou H. Case Report: A Deletion Variant in the DCAF17 Gene Underlying Woodhouse-Sakati Syndrome in a Chinese Consanguineous Family. Front Genet. 2021 Sep 23;12:741323. doi: 10.3389/fgene.2021.741323. eCollection 2021. Citation on PubMed
- Hdiji O, Turki E, Bouzidi N, Bouchhima I, Damak M, Bohlega S, Mhiri C. Woodhouse-Sakati Syndrome: Report of the First Tunisian Family with the C2orf37 Gene Mutation. J Mov Disord. 2016 May;9(2):120-3. doi: 10.14802/jmd.16003. Epub 2016 May 25. Citation on PubMed or Free article on PubMed Central
- Kohil A, Abdallah AM, Hussain K, Al-Shafai M. Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review. Orphanet J Rare Dis. 2023 Jan 31;18(1):22. doi: 10.1186/s13023-023-02614-8. Citation on PubMed
- Steindl K, Alazami AM, Bhatia KP, Wuerfel JT, Petersen D, Cartolari R, Neri G, Klein C, Mongiardo B, Alkuraya FS, Schneider SA. A novel C2orf37 mutation causes the first Italian cases of Woodhouse Sakati syndrome. Clin Genet. 2010 Dec;78(6):594-7. doi: 10.1111/j.1399-0004.2010.01447.x. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.