

Description
VEXAS syndrome is a disorder involving episodes of fever and abnormal inflammation. VEXAS is an acronym that stands for the technical terms of key descriptors of the condition. Normally, inflammation is an immune system response to injury or foreign invaders (such as bacteria). In people with VEXAS syndrome, part of the immune system called the innate immune response is turned on (activated) abnormally, when there is no injury or foreign invader, which causes fevers and inflammation-related damage to tissues and organs. Based on this process, VEXAS syndrome is classified as an autoinflammatory disease.
VEXAS syndrome typically affects older adults, primarily males, with signs and symptoms of the condition developing in a person's fifties, sixties, or seventies. People with VEXAS syndrome often have inflammation of the joints (arthritis), skin (dermatitis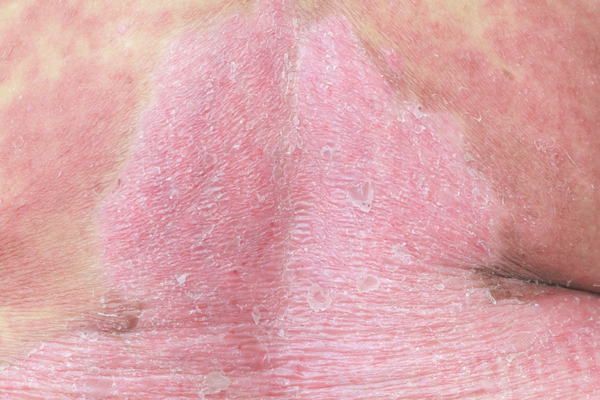 ), cartilage in the ear and nose (chondritis), or blood vessels (vasculitis
), cartilage in the ear and nose (chondritis), or blood vessels (vasculitis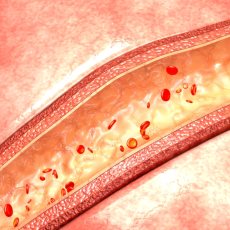 ). Inflammation can also develop in other tissues, including in the lungs and eyes. Affected individuals may also have enlarged lymph nodes
). Inflammation can also develop in other tissues, including in the lungs and eyes. Affected individuals may also have enlarged lymph nodes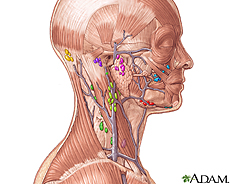 .
.
Blood cell abnormalities are common in VEXAS syndrome. Most affected individuals develop a shortage of red blood cells (a condition called anemia ), and the red blood cells that are present are abnormally large (macrocytic). People with VEXAS syndrome can also have a shortage of blood cells called platelets (a disorder known as thrombocytopenia); platelets
), and the red blood cells that are present are abnormally large (macrocytic). People with VEXAS syndrome can also have a shortage of blood cells called platelets (a disorder known as thrombocytopenia); platelets are needed for normal blood clotting. Some affected individuals develop myelodyspastic syndrome, a condition in which immature blood cells fail to develop normally; this condition may progress to a form of blood cancer called
leukemia
are needed for normal blood clotting. Some affected individuals develop myelodyspastic syndrome, a condition in which immature blood cells fail to develop normally; this condition may progress to a form of blood cancer called
leukemia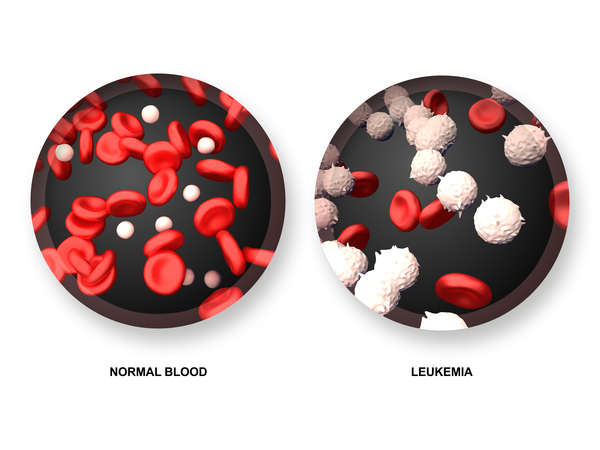 .
.
Frequency
VEXAS syndrome is a rare condition. Research suggests the condition affects an estimated 1 in 13,000 people.
Causes
Variants (also called mutations) in the UBA1 gene cause VEXAS syndrome. The UBA1 gene provides instructions for making a protein called ubiquitin-activating enzyme E1. This enzyme is necessary for a process that targets damaged or unneeded proteins to be broken down (degraded) within cells. Protein degradation helps maintain the proper balance of protein production and breakdown (protein homeostasis) that cells need to function and survive. The UBA1 gene variants that cause VEXAS syndrome are acquired during a person's lifetime and are present only in certain immune cells and blood-forming cells in the bone marrow . These changes, which are called somatic variants, are not inherited.
. These changes, which are called somatic variants, are not inherited.
UBA1 gene variants that cause VEXAS syndrome result in the production of an enzyme with reduced function. As a result, damaged or unneeded proteins build up inside cells instead of being broken down, which may contribute to abnormal activation of immune cells or cell damage and death. This protein buildup also disrupts protein homeostasis. Old proteins must be removed before cells can make new proteins. If damaged or unneeded proteins are not broken down, they can stop the production of new proteins, which can contribute to the impairment of normal cell functions. When UBA1 gene variants occur in immune cells or blood cells, they lead to abnormal inflammation, impaired blood cell development, and other features of VEXAS syndrome.
Inheritance
VEXAS syndrome is acquired, rather than inherited. Most cases result from new variants in the UBA1 gene, and occur in people with no previous history of the disorder in their family. The condition is not passed down to children of affected individuals.
The UBA1 gene is located on the X chromosome, which is one of the two sex chromosomes . Males have only one X chromosome, and a variant in the only copy of the UBA1 gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene, or there would have to be a loss of one of the X chromosomes, to cause the disorder.
. Males have only one X chromosome, and a variant in the only copy of the UBA1 gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene, or there would have to be a loss of one of the X chromosomes, to cause the disorder.
Other Names for This Condition
- vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome
- VEXAS
Additional Information & Resources
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Beck DB, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, Magaziner SJ, Strande NT, Cantor A, Haley JS, Cook A, Hill W, Schwartz AL, Grayson PC, Ferrada MA, Kastner DL, Carey DJ, Stewart DR. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated With VEXAS Syndrome in a Clinical Population. JAMA. 2023 Jan 24;329(4):318-324. doi: 10.1001/jama.2022.24836. Citation on PubMed
- Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, Balanda N, Ross DL, Ospina Cardona D, Wu Z, Patel B, Manthiram K, Groarke EM, Gutierrez-Rodrigues F, Hoffmann P, Rosenzweig S, Nakabo S, Dillon LW, Hourigan CS, Tsai WL, Gupta S, Carmona-Rivera C, Asmar AJ, Xu L, Oda H, Goodspeed W, Barron KS, Nehrebecky M, Jones A, Laird RS, Deuitch N, Rowczenio D, Rominger E, Wells KV, Lee CR, Wang W, Trick M, Mullikin J, Wigerblad G, Brooks S, Dell'Orso S, Deng Z, Chae JJ, Dulau-Florea A, Malicdan MCV, Novacic D, Colbert RA, Kaplan MJ, Gadina M, Savic S, Lachmann HJ, Abu-Asab M, Solomon BD, Retterer K, Gahl WA, Burgess SM, Aksentijevich I, Young NS, Calvo KR, Werner A, Kastner DL, Grayson PC. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020 Dec 31;383(27):2628-2638. doi: 10.1056/NEJMoa2026834. Epub 2020 Oct 27. Citation on PubMed
- Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, Lacombe V, Terriou L, Ardois S, Bouaziz JD, Mathian A, Le Guenno G, Aouba A, Outh R, Meyer A, Roux-Sauvat M, Ebbo M, Zhao LP, Bigot A, Jamilloux Y, Guillotin V, Flamarion E, Henneton P, Vial G, Jachiet V, Rossignol J, Vinzio S, Weitten T, Vinit J, Deligny C, Humbert S, Samson M, Magy-Bertrand N, Moulinet T, Bourguiba R, Hanslik T, Bachmeyer C, Sebert M, Kostine M, Bienvenu B, Biscay P, Liozon E, Sailler L, Chasset F, Audemard-Verger A, Duroyon E, Sarrabay G, Borlot F, Dieval C, Cluzeau T, Marianetti P, Lobbes H, Boursier G, Gerfaud-Valentin M, Jeannel J, Servettaz A, Audia S, Larue M, Henriot B, Faucher B, Graveleau J, de Sainte Marie B, Galland J, Bouillet L, Arnaud C, Ades L, Carrat F, Hirsch P, Fenaux P, Fain O, Sujobert P, Kosmider O, Mekinian A; French VEXAS group; GFEV, GFM, CEREMAIA, MINHEMON. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. 2022 Mar;186(3):564-574. doi: 10.1111/bjd.20805. Epub 2021 Nov 28. Citation on PubMed
- Koster MJ, Kourelis T, Reichard KK, Kermani TA, Beck DB, Cardona DO, Samec MJ, Mangaonkar AA, Begna KH, Hook CC, Oliveira JL, Nasr SH, Tiong BK, Patnaik MM, Burke MM, Michet CJ Jr, Warrington KJ. Clinical Heterogeneity of the VEXAS Syndrome: A Case Series. Mayo Clin Proc. 2021 Oct;96(10):2653-2659. doi: 10.1016/j.mayocp.2021.06.006. Epub 2021 Sep 3. Citation on PubMed
- Muratore F, Marvisi C, Castrignano P, Nicoli D, Farnetti E, Bonanno O, Longo R, Zaldini P, Galli E, Balanda N, Beck DB, Grayson PC, Pipitone N, Boiardi L, Salvarani C. VEXAS Syndrome: A Case Series From a Single-Center Cohort of Italian Patients With Vasculitis. Arthritis Rheumatol. 2022 Apr;74(4):665-670. doi: 10.1002/art.41992. Epub 2022 Mar 3. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.