

Description
Tyrosinemia is a genetic disorder characterized by problems breaking down the amino acid tyrosine, which is a building block of most proteins. If the condition is untreated, tyrosine and its byproducts build up in tissues and organs, which can lead to serious health problems.
tyrosine, which is a building block of most proteins. If the condition is untreated, tyrosine and its byproducts build up in tissues and organs, which can lead to serious health problems.
There are three types of tyrosinemia, distinguished by their symptoms and genetic cause. Tyrosinemia type I is the most severe form of this disorder and usually begins in the first few months of life. Affected infants do not gain weight and grow at the expected rate (failure to thrive) because eating high-protein foods leads to diarrhea and vomiting. Affected infants may also have yellowing of the skin and whites of the eyes (jaundice), a cabbage-like odor, and an increased tendency to bleed (particularly nosebleeds).
In addition, tyrosinemia type I can lead to liver and kidney failure, softening and weakening of the bones (rickets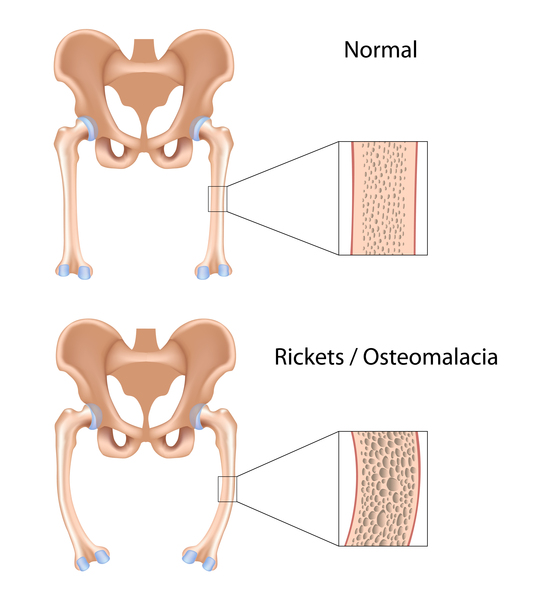 ), and an increased risk of liver cancer
), and an increased risk of liver cancer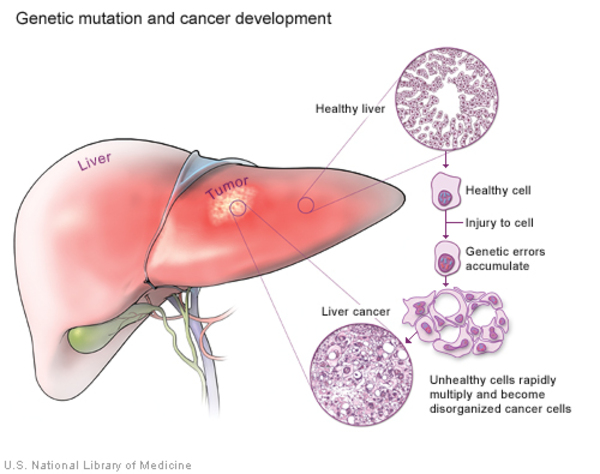 (hepatocellular carcinoma). Some affected children have repeated neurologic crises that consist of changes in their mental state, reduced sensation in the arms and legs (peripheral neuropathy), abdominal pain, and serious breathing problems (respiratory failure). These crises can last from 1 to 7 days. Without treatment, children with tyrosinemia type I often do not survive past the age of 10. With early diagnosis and treatment, though, affected individuals can live into adulthood.
(hepatocellular carcinoma). Some affected children have repeated neurologic crises that consist of changes in their mental state, reduced sensation in the arms and legs (peripheral neuropathy), abdominal pain, and serious breathing problems (respiratory failure). These crises can last from 1 to 7 days. Without treatment, children with tyrosinemia type I often do not survive past the age of 10. With early diagnosis and treatment, though, affected individuals can live into adulthood.
Tyrosinemia type II often begins in early childhood and affects the eyes, skin, and mental development. Signs and symptoms include eye pain and redness, excessive tearing, abnormal sensitivity to light (photophobia), and thick, painful skin on the palms of the hands and soles of the feet (palmoplantar hyperkeratosis). About half of individuals with tyrosinemia type II have some degree of intellectual disability.
Tyrosinemia type III is the rarest of the three types. The characteristic features of this type include intellectual disabilities, seizures, and periodic loss of balance and coordination (intermittent ataxia). Liver problems do not occur in types II and III.
About 1 in 10 of all newborns have temporarily elevated levels of tyrosine (transient tyrosinemia). These cases are not genetic. The most likely causes are vitamin C deficiency or an immature liver due to premature birth.
Frequency
Worldwide, tyrosinemia type I affects about 1 in 100,000 individuals. This type is more common in Norway, where 1 in 60,000 to 74,000 individuals are affected, and Quebec, Canada, where it occurs in about 1 in 16,000 individuals. It is especially common in the Saguenay-Lac St. Jean region of Quebec, where it affects 1 in 1,846 people.
Tyrosinemia type II occurs in fewer than 1 in 250,000 individuals worldwide.
Tyrosinemia type III is very rare; only a few cases have been reported.
Causes
Variants (also called mutations) in the FAH, TAT, and HPD genes can cause tyrosinemia types I, II, and III, respectively.
In the liver , specialized proteins called enzymes break down tyrosine in a five-step process. The resulting molecules are either removed from the body by the kidneys
, specialized proteins called enzymes break down tyrosine in a five-step process. The resulting molecules are either removed from the body by the kidneys or used in the body to produce energy or make other substances.
or used in the body to produce energy or make other substances.
The FAH gene provides instructions for making an enzyme called fumarylacetoacetate hydrolase, which performs the last step of tyrosine breakdown. The enzyme produced from the TAT gene, called tyrosine aminotransferase enzyme, is involved in the first step in the process. The HPD gene provides instructions for making the 4-hydroxyphenylpyruvate dioxygenase enzyme, which performs the second step.
Variants in the FAH, TAT, or HPD gene reduce the activity of one of the enzymes that break down tyrosine. As a result, tyrosine and its byproducts accumulate to toxic levels, which can damage and kill cells in the liver, kidneys, nervous system, and other organs.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Hereditary tyrosinemia
- Hypertyrosinaemia
- Hypertyrosinemia
- Tyrosinaemia
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bouyacoub Y, Zribi H, Azzouz H, Nasrallah F, Abdelaziz RB, Kacem M, Rekaya B, Messaoud O, Romdhane L, Charfeddine C, Bouziri M, Bouziri S, Tebib N, Mokni M, Kaabachi N, Boubaker S, Abdelhak S. Novel and recurrent mutations in the TAT gene in Tunisian families affected with Richner-Hanhart syndrome. Gene. 2013 Oct 15;529(1):45-9. doi: 10.1016/j.gene.2013.07.066. Epub 2013 Aug 13. Citation on PubMed
- Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CDM, Grompe M, Mitchell G, Waisbren SE, Gucsavas-Calikoglu M, Wasserstein MP, Coakley K, Scott CR. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med. 2017 Dec;19(12). doi: 10.1038/gim.2017.101. Epub 2017 Aug 3. Citation on PubMed
- Couce ML, Dalmau J, del Toro M, Pintos-Morell G, Aldamiz-Echevarria L; Spanish Working Group on Tyrosinemia type 1. Tyrosinemia type 1 in Spain: mutational analysis, treatment and long-term outcome. Pediatr Int. 2011 Dec;53(6):985-9. doi: 10.1111/j.1442-200X.2011.03427.x. Citation on PubMed
- de Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L, de Baulny HO, Pintos-Morell G, Spiekerkotter U. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis. 2013 Jan 11;8:8. doi: 10.1186/1750-1172-8-8. Citation on PubMed or Free article on PubMed Central
- Fernandez-Lainez C, Ibarra-Gonzalez I, Belmont-Martinez L, Monroy-Santoyo S, Guillen-Lopez S, Vela-Amieva M. Tyrosinemia type I: clinical and biochemical analysis of patients in Mexico. Ann Hepatol. 2014 Mar-Apr;13(2):265-72. Citation on PubMed
- Ficicioglu C. Tyrosinemia Type I. 2006 Jul 24 [updated 2025 Nov 20]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1515/ Citation on PubMed
- Heylen E, Scherer G, Vincent MF, Marie S, Fischer J, Nassogne MC. Tyrosinemia Type III detected via neonatal screening: management and outcome. Mol Genet Metab. 2012 Nov;107(3):605-7. doi: 10.1016/j.ymgme.2012.09.002. Epub 2012 Sep 7. Citation on PubMed
- Macsai MS, Schwartz TL, Hinkle D, Hummel MB, Mulhern MG, Rootman D. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001 Oct;132(4):522-7. doi: 10.1016/s0002-9394(01)01160-6. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.