

Description
Trichorhinophalangeal syndrome type II (TRPS II) is a condition that causes bone and joint malformations; distinctive facial features; intellectual disability; and abnormalities of the skin, hair, teeth, sweat glands, and nails. The name of the condition describes some of the areas of the body that are commonly affected: hair (tricho-), nose (rhino-), and fingers and toes (phalangeal).
People with this condition have multiple noncancerous (benign) bone tumors called osteochondromas. Affected individuals may develop a few to several hundred osteochondromas. These bone growths typically begin in infancy to early childhood and stop forming around adolescence. Depending on the location of the osteochondromas, they can cause pain, limited range of joint movement, or damage to blood vessels or the spinal cord. Individuals with TRPS II may have reduced bone mineral density (osteopenia). Affected individuals often have slow growth before and after birth resulting in short stature. In TRPS II, the ends (epiphyses) of one or more bones in the fingers or toes are abnormally cone-shaped. Additionally, the fingernails and toenails are typically thin and abnormally formed.
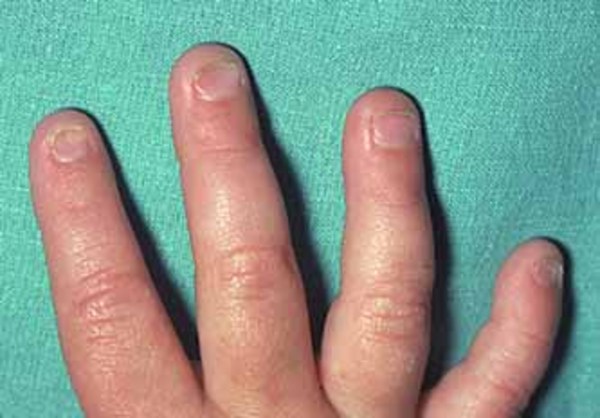
Children with TRPS II often have an unusually large range of joint movement (hypermobility). However, as osteochondromas begin to develop, typically starting between infancy and mid-childhood, the joints begin to stiffen, leading to decreased mobility. Individuals with TRPS II may also have a misalignment of the hip joints (hip dysplasia), which often develops in early adulthood but can occur in infancy or childhood.

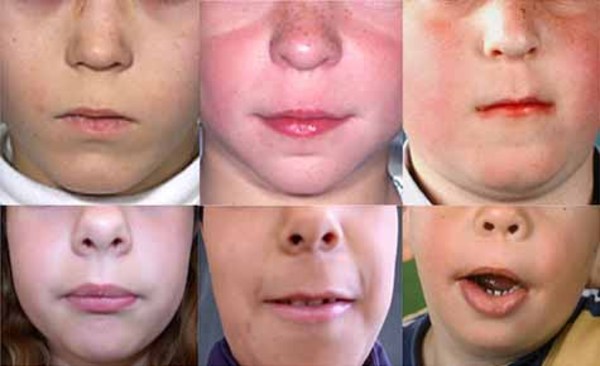
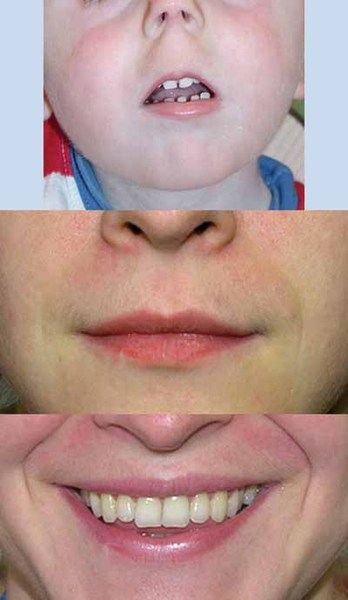
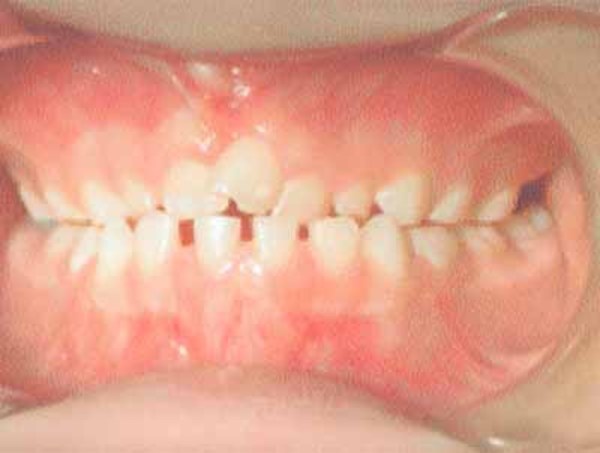
The characteristic appearance of individuals with TRPS II involves thick eyebrows; a broad nose with a rounded tip; a long, smooth area between the nose and the upper lip (philtrum); a thin upper lip; and small teeth that are either decreased (oligodontia) or increased (supernumerary) in number. Almost all affected individuals have sparse scalp hair. Males are particularly affected by hair loss, with many being nearly or completely bald soon after puberty. Some children with this condition have loose skin, but the skin becomes tighter over time. Individuals with TRPS II may experience excessive sweating (hyperhidrosis).
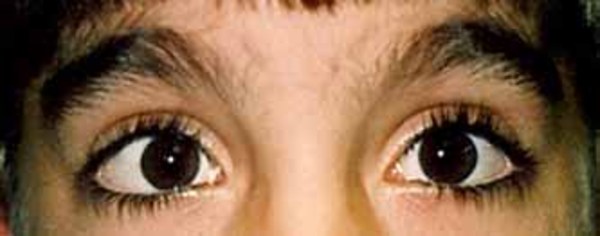
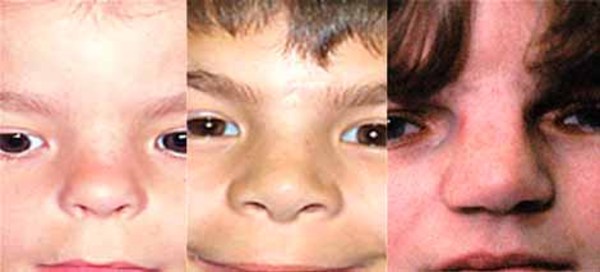
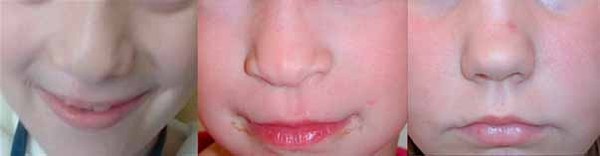


Most individuals with TRPS II have mild intellectual disability.
Frequency
TRPS II is a rare condition; its prevalence is unknown.
Causes
TRPS II is caused by the deletion of genetic material on the long arm (q) of chromosome 8. The size of the deletion varies among affected individuals; studies suggest that larger deletions tend to result in a greater number of features than do smaller deletions.
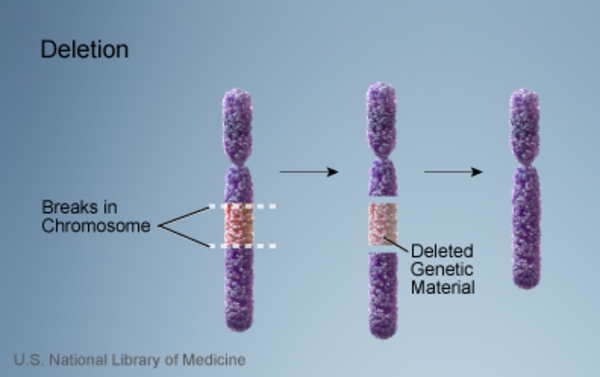
The signs and symptoms of TRPS II are related to the loss of multiple genes on chromosome 8. The TRPS1, EXT1, and RAD21 genes are missing in people with TRPS II. These genes play significant roles in regulating gene activity, protein function, and cell division.
Researchers have determined that the loss of the EXT1 gene is responsible for the multiple osteochondromas seen in people with TRPS II. Loss of the TRPS1 gene is thought to cause the other bone and facial abnormalities. Deletion of the RAD21 gene may contribute to intellectual disability. The loss of other genes from this region of chromosome 8 likely contributes to the additional features of this condition.
TRPS II is often described as a contiguous gene deletion syndrome because it results from the loss of several neighboring genes.
A condition similar to TRPS II is caused by gene changes that affect only the TRPS1 gene. This condition, called trichorhinophalangeal syndrome type I (TRPS I), features similar bone, joint, skin, and facial characteristics as TRPS II. Individuals with TRPSI do not have osteochondromas or intellectual disability, which are not associated with the TRPS1 gene.
Inheritance
Most cases of TRPS II are not inherited, but occur as random events during the formation of reproductive cells (eggs or sperm) in a parent of an affected individual. These cases occur in people with no history of the disorder in their family. In a very small number of cases, people with TRPS II have inherited the chromosomal deletion from a parent with the condition.

TRPS II is considered an autosomal dominant condition because one copy of the altered chromosome 8 in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Chromosome 8q24.1 deletion syndrome
- Giedion-Langer syndrome
- Langer-Giedion syndrome
- LGS
- Tricho-rhino-phalangeal syndrome type II
- Trichorhinophalangeal syndrome with exostosis
- TRPS II
- TRPS2
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Hilton MJ, Sawyer JM, Gutierrez L, Hogart A, Kung TC, Wells DE. Analysis of novel and recurrent mutations responsible for the tricho-rhino-phalangeal syndromes. J Hum Genet. 2002;47(3):103-6. doi: 10.1007/s100380200010. Citation on PubMed
- Maas SM, Shaw AC, Bikker H, Ludecke HJ, van der Tuin K, Badura-Stronka M, Belligni E, Biamino E, Bonati MT, Carvalho DR, Cobben J, de Man SA, Den Hollander NS, Di Donato N, Garavelli L, Gronborg S, Herkert JC, Hoogeboom AJ, Jamsheer A, Latos-Bielenska A, Maat-Kievit A, Magnani C, Marcelis C, Mathijssen IB, Nielsen M, Otten E, Ousager LB, Pilch J, Plomp A, Poke G, Poluha A, Posmyk R, Rieubland C, Silengo M, Simon M, Steichen E, Stumpel C, Szakszon K, Polonkai E, van den Ende J, van der Steen A, van Essen T, van Haeringen A, van Hagen JM, Verheij JB, Mannens MM, Hennekam RC. Phenotype and genotype in 103 patients with tricho-rhino-phalangeal syndrome. Eur J Med Genet. 2015 May;58(5):279-92. doi: 10.1016/j.ejmg.2015.03.002. Epub 2015 Mar 16. Citation on PubMed
- Riedl S, Giedion A, Schweitzer K, Mullner-Eidenbock A, Grill F, Frisch H, Ludecke HJ. Pronounced short stature in a girl with tricho-rhino-phalangeal syndrome II (TRPS II, Langer-Giedion syndrome) and growth hormone deficiency. Am J Med Genet A. 2004 Dec 1;131(2):200-3. doi: 10.1002/ajmg.a.30374. Citation on PubMed
- Schinzel A, Riegel M, Baumer A, Superti-Furga A, Moreira LM, Santo LD, Schiper PP, Carvalho JH, Giedion A. Long-term follow-up of four patients with Langer-Giedion syndrome: clinical course and complications. Am J Med Genet A. 2013 Sep;161A(9):2216-25. doi: 10.1002/ajmg.a.36062. Epub 2013 Aug 2. Citation on PubMed
- Shanske AL, Patel A, Saukam S, Levy B, Ludecke HJ. Clinical and molecular characterization of a patient with Langer-Giedion syndrome and mosaic del(8)(q22.3q24.13). Am J Med Genet A. 2008 Dec 15;146A(24):3211-6. doi: 10.1002/ajmg.a.32615. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.