

Description
Stüve-Wiedemann syndrome is a severe condition characterized by bone abnormalities and dysfunction of the autonomic nervous system, which controls involuntary body processes such as the regulation of breathing rate and body temperature. The condition is apparent from birth, and its key features include abnormal curvature (bowing) of the long bones in the legs, difficulty feeding and swallowing, and episodes of dangerously high body temperature (hyperthermia).
In addition to bowed legs, affected infants can have bowed arms, permanently bent fingers and toes (camptodactyly), and joint deformities (contractures) in the elbows and knees that restrict their movement. Other features include abnormalities of the pelvic bones (the ilia) and reduced bone mineral density (osteopenia).
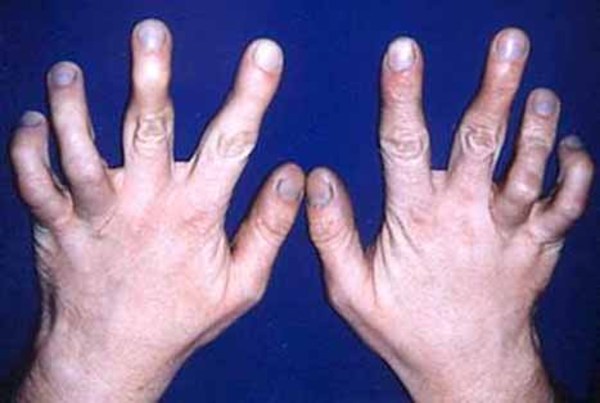
In infants with Stüve-Wiedemann syndrome, dysfunction of the autonomic nervous system typically leads to difficulty feeding and swallowing, breathing problems, and episodes of hyperthermia. Affected infants may also sweat excessively, even when the body temperature is not elevated, or have a reduced ability to feel pain. Many babies with this condition do not survive past infancy because of the problems regulating breathing and body temperature; however, some people with Stüve-Wiedemann syndrome live into adolescence or later.
Problems with breathing and swallowing usually improve in affected children who survive infancy; however, they still have difficulty regulating body temperature. In addition, the leg bowing worsens, and children with Stüve-Wiedemann syndrome may develop prominent joints, an abnormal curvature of the spine (scoliosis), and spontaneous bone fractures. Some affected individuals have a smooth tongue that lacks the bumps that house taste buds (fungiform papillae). Affected children may also lose certain reflexes, particularly the reflex to blink when something touches the eye (corneal reflex) and the knee-jerk reflex (patellar reflex).


Another condition once known as Schwartz-Jampel syndrome type 2 is now considered to be part of Stüve-Wiedemann syndrome. Researchers have recommended that the designation Schwartz-Jampel syndrome type 2 no longer be used.
Frequency
Stüve-Wiedemann syndrome is a rare condition that has been found worldwide. Its prevalence is unknown.
Causes
Stüve-Wiedemann syndrome is usually caused by mutations in the LIFR gene. This gene provides instructions for making a protein called leukemia inhibitory factor receptor (LIFR). Receptor proteins have specific sites into which certain other proteins, called ligands, fit like keys into locks. Together, ligands and their receptors trigger signals that affect cell development and function.
The LIFR protein acts as a receptor for a ligand known as leukemia inhibitory factor (LIF). LIFR signaling can control several cellular processes, including growth and division (proliferation), maturation (differentiation), and survival. First found to be important in blocking (inhibiting) growth of blood cancer (leukemia) cells, this signaling is also involved in the formation of bone and the development of nerve cells. It appears to play an important role in normal development and functioning of the autonomic nervous system.
Most LIFR gene mutations that cause Stüve-Wiedemann syndrome prevent production of any LIFR protein. Other mutations lead to production of an altered protein that likely cannot function. Without functional LIFR, signaling is impaired. The lack of LIFR signaling disrupts normal bone formation, leading to osteopenia, bowed legs, and other skeletal problems common in Stüve-Wiedemann syndrome. In addition, development of nerve cells, particularly those involved in the autonomic nervous system, is abnormal, leading to the problems with breathing, feeding, and regulating body temperature characteristic of this condition.
A small number of people with Stüve-Wiedemann syndrome do not have an identified mutation in the LIFR gene. Researchers suggest that other genes that have not been identified may be involved in this condition.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Neonatal Schwartz-Jampel syndrome
- Schwartz-Jampel type 2 syndrome
- SJS2
- Stuve-Wiedemann dysplasia
- Stuve-Wiedemann syndrome
- Stuve-Wiedemann/Schwartz-Jampel type 2 syndrome
- STWS
- SWS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dagoneau N, Scheffer D, Huber C, Al-Gazali LI, Di Rocco M, Godard A, Martinovic J, Raas-Rothschild A, Sigaudy S, Unger S, Nicole S, Fontaine B, Taupin JL, Moreau JF, Superti-Furga A, Le Merrer M, Bonaventure J, Munnich A, Legeai-Mallet L, Cormier-Daire V. Null leukemia inhibitory factor receptor (LIFR) mutations in Stuve-Wiedemann/Schwartz-Jampel type 2 syndrome. Am J Hum Genet. 2004 Feb;74(2):298-305. doi: 10.1086/381715. Epub 2004 Jan 21. Citation on PubMed or Free article on PubMed Central
- Jung C, Dagoneau N, Baujat G, Le Merrer M, David A, Di Rocco M, Hamel B, Megarbane A, Superti-Furga A, Unger S, Munnich A, Cormier-Daire V. Stuve-Wiedemann syndrome: long-term follow-up and genetic heterogeneity. Clin Genet. 2010 Mar;77(3):266-72. doi: 10.1111/j.1399-0004.2009.01314.x. Citation on PubMed
- Mikelonis D, Jorcyk CL, Tawara K, Oxford JT. Stuve-Wiedemann syndrome: LIFR and associated cytokines in clinical course and etiology. Orphanet J Rare Dis. 2014 Mar 12;9:34. doi: 10.1186/1750-1172-9-34. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.