

Description
Spastic paraplegia type 49 is part of a group of genetic disorders known as hereditary spastic paraplegias. These disorders are characterized by progressive muscle stiffness (spasticity) and the development of paralysis of the lower limbs (paraplegia). Hereditary spastic paraplegias are divided into two types: pure and complex. The pure types involve only the lower limbs, whereas the complex types also involve the upper limbs (to a lesser degree) and other problems with the nervous system. Spastic paraplegia type 49 is a complex hereditary spastic paraplegia.
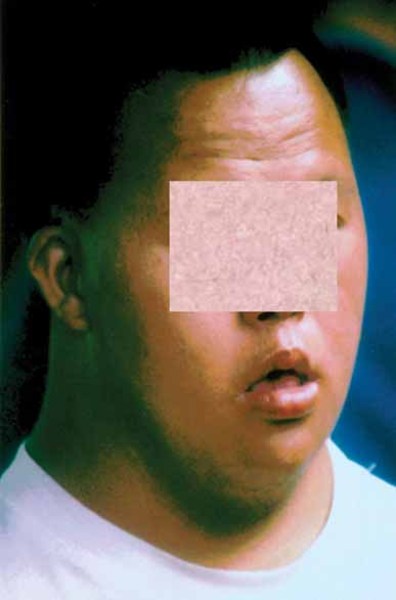
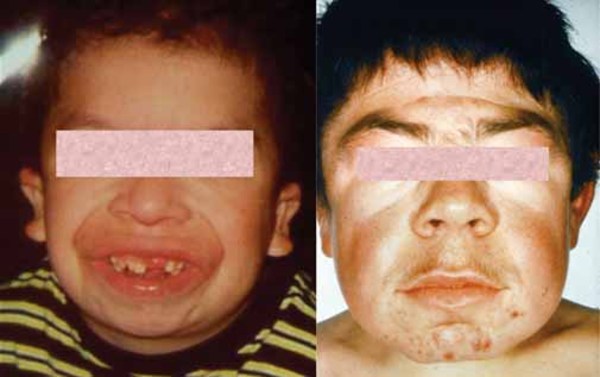
Spastic paraplegia type 49 often begins with weak muscle tone (hypotonia) that starts in infancy. During childhood, spasticity and paraplegia develop and gradually worsen, causing difficulty walking and frequent falls. In addition, affected individuals have moderate to severe intellectual disability and distinctive physical features, including short stature; chubbiness; an unusually small head size (microcephaly); a wide, short skull (brachycephaly); a short, broad neck; and facial features described as coarse. Some people with spastic paraplegia type 49 develop seizures.


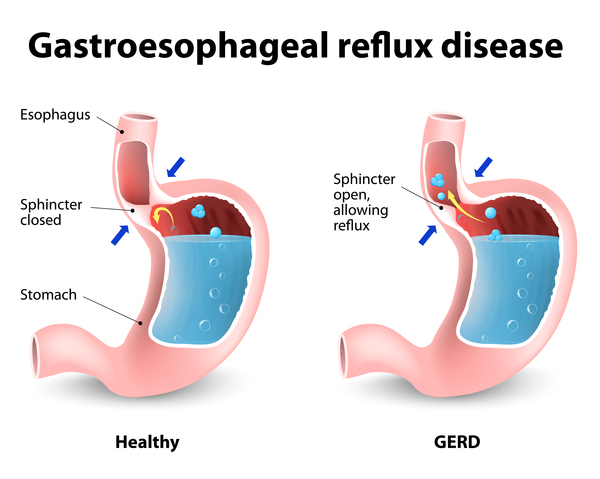
Problems with autonomic nerve cells (autonomic neurons), which control involuntary body functions such as heart rate, digestion, and breathing, result in several features of spastic paraplegia type 49. Affected individuals have difficulty feeding beginning in infancy. They experience a backflow of stomach acids into the esophagus (called gastroesophageal reflux or GERD), causing vomiting. GERD can also lead to recurrent bacterial lung infections called aspiration pneumonia, which can be life-threatening. In addition, people with spastic paraplegia type 49 have problems regulating their breathing, resulting in pauses in breathing (apnea), initially while sleeping but eventually also while awake. Their blood pressure, pulse rate, and body temperature are also irregular.
People with spastic paraplegia type 49 can develop recurrent episodes of severe weakness, hypotonia, and abnormal breathing, which can be life threatening. By early adulthood, some affected individuals need a machine to help them breathe (mechanical ventilation).
Other signs and symptoms of spastic paraplegia type 49 reflect problems with sensory neurons, which transmit information about sensations such as pain, temperature, and touch to the brain. Many affected individuals are less able to feel pain or temperature sensations than individuals in the general population. Affected individuals also have abnormal or absent reflexes (areflexia).
Because of the nervous system abnormalities that occur in spastic paraplegia type 49, it has been suggested that the condition also be classified as a hereditary sensory and autonomic neuropathy, which is a group of conditions that affect sensory and autonomic neurons.
Frequency
Spastic paraplegia type 49 is a rare disorder. Its prevalence is unknown.
Causes
Spastic paraplegia type 49 is caused by mutations in the TECPR2 gene. The protein produced from this gene plays a role in a cellular process called autophagy, by which worn-out or unnecessary cell parts are broken down and recycled. During autophagy, materials that are no longer needed are isolated in compartments called autophagosomes and transported to cell structures that break them down. The TECPR2 protein is thought to be important for the formation of autophagosomes.
The TECPR2 gene mutations that cause spastic paraplegia type 49 likely result in an abnormal or absent TECPR2 protein. Alteration or loss of this protein is thought to impair autophagy, making cells less efficient at removing unneeded materials. Researchers suggest that neurons may be particularly vulnerable to impaired autophagy because it is especially difficult to transport waste materials through their long extensions (axons and dendrites) for breakdown. The waste materials can build up in neurons and damage them. Damage to autonomic and sensory neurons and neurons that control movement (motor neurons) results in the signs and symptoms of spastic paraplegia type 49.

Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Autosomal recessive spastic paraplegia type 49
- Spastic paraplegia 49, autosomal recessive
- SPG49
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Covone AE, Fiorillo C, Acquaviva M, Trucco F, Morana G, Ravazzolo R, Minetti C. WES in a family trio suggests involvement of TECPR2 in a complex form of progressive motor neuron disease. Clin Genet. 2016 Aug;90(2):182-5. doi: 10.1111/cge.12730. Epub 2016 Feb 10. Citation on PubMed
- Heimer G, Oz-Levi D, Eyal E, Edvardson S, Nissenkorn A, Ruzzo EK, Szeinberg A, Maayan C, Mai-Zahav M, Efrati O, Pras E, Reznik-Wolf H, Lancet D, Goldstein DB, Anikster Y, Shalev SA, Elpeleg O, Ben Zeev B. TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur J Paediatr Neurol. 2016 Jan;20(1):69-79. doi: 10.1016/j.ejpn.2015.10.003. Epub 2015 Oct 22. Citation on PubMed
- Oz-Levi D, Ben-Zeev B, Ruzzo EK, Hitomi Y, Gelman A, Pelak K, Anikster Y, Reznik-Wolf H, Bar-Joseph I, Olender T, Alkelai A, Weiss M, Ben-Asher E, Ge D, Shianna KV, Elazar Z, Goldstein DB, Pras E, Lancet D. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet. 2012 Dec 7;91(6):1065-72. doi: 10.1016/j.ajhg.2012.09.015. Epub 2012 Nov 21. Citation on PubMed or Free article on PubMed Central
- Stadel D, Millarte V, Tillmann KD, Huber J, Tamin-Yecheskel BC, Akutsu M, Demishtein A, Ben-Zeev B, Anikster Y, Perez F, Dotsch V, Elazar Z, Rogov V, Farhan H, Behrends C. TECPR2 Cooperates with LC3C to Regulate COPII-Dependent ER Export. Mol Cell. 2015 Oct 1;60(1):89-104. doi: 10.1016/j.molcel.2015.09.010. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.