

Description
Snijders Blok-Campeau syndrome is characterized by intellectual disability, speech problems, and distinctive facial features.
Intellectual disability in individuals with Snijders Blok-Campeau syndrome ranges from mild to severe. Some people with this condition also have low muscle tone (hypotonia), seizures, or autistic behaviors that affect communication and social interaction.
While some people with Snijders Blok-Campeau syndrome develop limited language, others acquire only a few words or never speak. If speech occurs, it usually develops after age 2. Affected individuals can experience stuttering, problems coordinating movements of the mouth and tongue (oromotor dysfunction), or difficulty producing the sequences of sounds and syllables needed to form words (apraxia). In general, people with this condition have a very social personality.
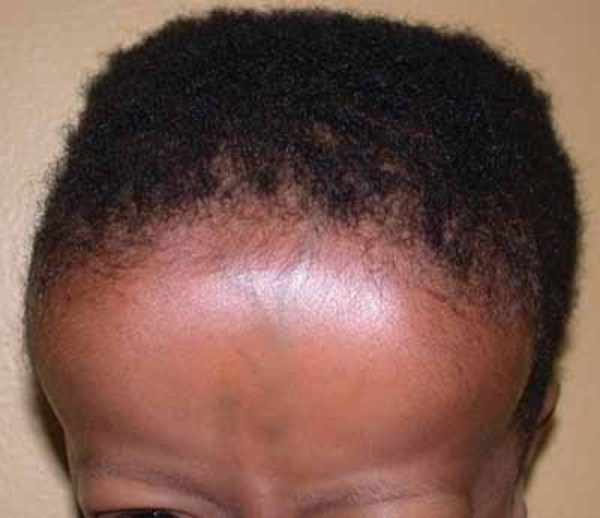
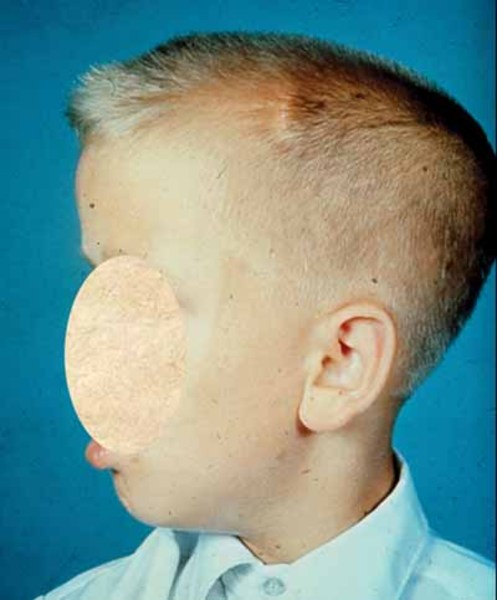
Individuals with Snijders Blok-Campeau syndrome have distinctive facial features. The eyes are frequently affected, and features often include widely spaced eyes (ocular hypertelorism), deep-set eyes, narrowed openings of the eyes (narrowed palpebral fissures), an increased distance between the inner corners of the eyes (telecanthus), and sparse eyebrows. Additional facial features can include full cheeks, a pointed chin, a prominent forehead (frontal bossing), a sunken appearance of the middle of the face (midface hypoplasia), a broad nasal bridge, low-set ears that may be rotated backward, and a thin upper lip. Affected individuals often have an abnormally sized head; most have an unusually large head (macrocephaly), though some have an unusually small head (microcephaly). Some people with Snijders Blok-Campeau syndrome have premature closure of certain bones of the skull (craniosynostosis), which can contribute to an abnormal head shape.

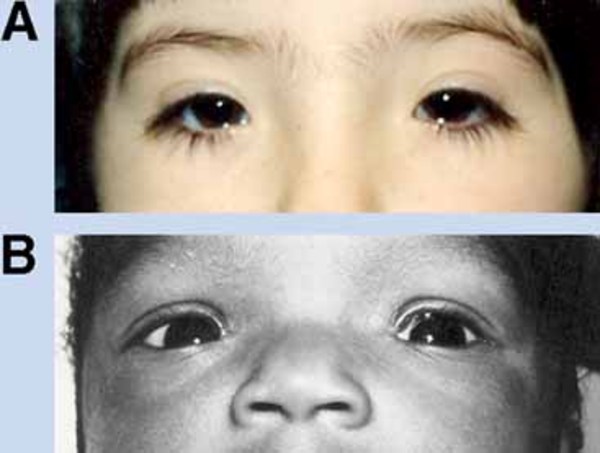
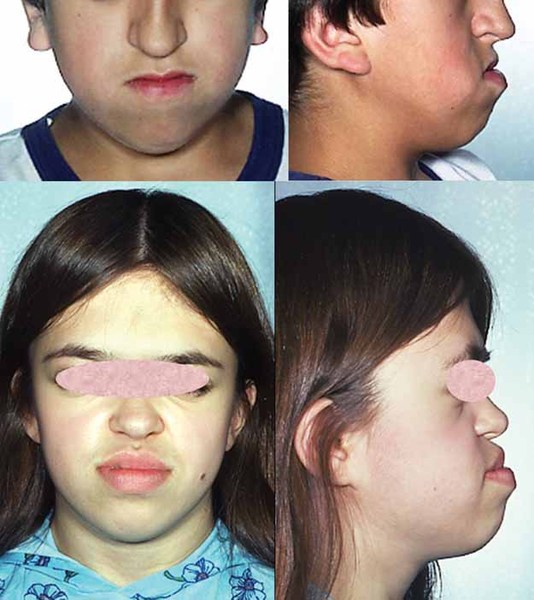

Most individuals with Snijders Blok-Campeau syndrome have vision problems, including farsightedness (hyperopia) or eyes that do not look in the same direction (strabismus).
About half of people with Snijders Blok-Campeau syndrome have brain abnormalities, such as enlarged spaces in the brain where cerebrospinal fluid (CSF) accumulates. Less commonly, affected individuals are born with a hole between the two upper chambers of the heart (atrial septal defect) or another problem with the heart's structure or function (congenital heart disease).

Frequency
The prevalence of Snijders Blok-Campeau syndrome is unknown. It is thought to be a rare condition. Approximately 60 cases have been described in the scientific literature.
Causes
Snijders Blok-Campeau syndrome is caused by mutations in the CHD3 gene. This gene provides instructions for making a protein that regulates gene activity (expression) by a process known as chromatin remodeling. Chromatin is the network of DNA and proteins that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly DNA is packaged. Chromatin remodeling is one way gene expression is regulated during development; when DNA is tightly packed, gene expression is lower than when DNA is loosely packed. Through its ability to regulate gene activity, the CHD3 protein is involved in many processes during development, including maintenance of the structure and integrity of DNA, cell growth and division (proliferation), and the maturation (differentiation) of cells such as nerve cells (neurons).
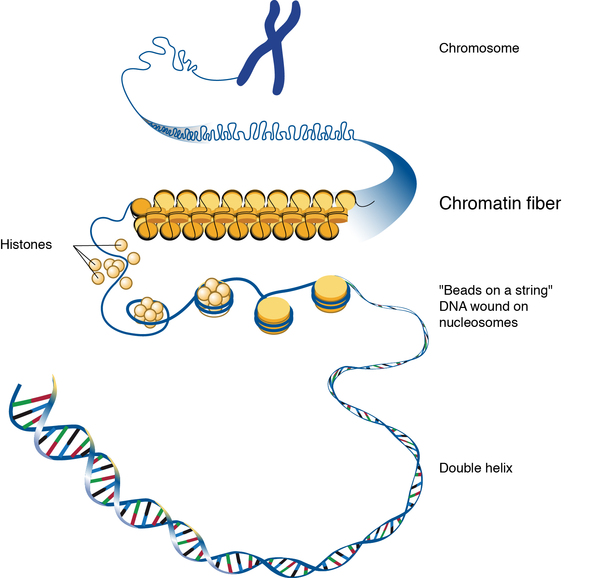
Some CHD3 gene mutations increase the function of the protein, while most reduce the protein's activity. It is likely that either an increase or a decrease in CHD3 protein activity alters chromatin remodeling, which disrupts normal gene expression. Changes in CHD3 protein activity seem to affect the activity of genes that direct the development of many different organs and tissues before birth. It is unclear how increased and decreased protein function both lead to the signs and symptoms of Snijders Blok-Campeau syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Most cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual’s parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- IDDMSF
- Intellectual developmental disorder with macrocephaly, speech delay, and dysmorphic facies
- SNIBCPS
Additional Information & Resources
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Hoffmann A, Spengler D. Chromatin Remodeling Complex NuRD in Neurodevelopment and Neurodevelopmental Disorders. Front Genet. 2019 Jul 24;10:682. doi: 10.3389/fgene.2019.00682. eCollection 2019. Citation on PubMed or Free article on PubMed Central
- Pierson TM, Otero MG, Grand K, Choi A, Graham JM Jr, Young JI, Mackay JP. The NuRD complex and macrocephaly associated neurodevelopmental disorders. Am J Med Genet C Semin Med Genet. 2019 Dec;181(4):548-556. doi: 10.1002/ajmg.c.31752. Epub 2019 Nov 18. Citation on PubMed
- Snijders Blok L, Rousseau J, Twist J, Ehresmann S, Takaku M, Venselaar H, Rodan LH, Nowak CB, Douglas J, Swoboda KJ, Steeves MA, Sahai I, Stumpel CTRM, Stegmann APA, Wheeler P, Willing M, Fiala E, Kochhar A, Gibson WT, Cohen ASA, Agbahovbe R, Innes AM, Au PYB, Rankin J, Anderson IJ, Skinner SA, Louie RJ, Warren HE, Afenjar A, Keren B, Nava C, Buratti J, Isapof A, Rodriguez D, Lewandowski R, Propst J, van Essen T, Choi M, Lee S, Chae JH, Price S, Schnur RE, Douglas G, Wentzensen IM, Zweier C, Reis A, Bialer MG, Moore C, Koopmans M, Brilstra EH, Monroe GR, van Gassen KLI, van Binsbergen E, Newbury-Ecob R, Bownass L, Bader I, Mayr JA, Wortmann SB, Jakielski KJ, Strand EA, Kloth K, Bierhals T; DDD study; Roberts JD, Petrovich RM, Machida S, Kurumizaka H, Lelieveld S, Pfundt R, Jansen S, Deriziotis P, Faivre L, Thevenon J, Assoum M, Shriberg L, Kleefstra T, Brunner HG, Wade PA, Fisher SE, Campeau PM. CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat Commun. 2018 Nov 5;9(1):4619. doi: 10.1038/s41467-018-06014-6. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.