

Description
Scalp-ear-nipple syndrome, as its name suggests, is a condition characterized by abnormalities of the scalp, ears, and nipples. Less frequently, affected individuals have problems affecting other parts of the body. The features of this disorder can vary even within the same family.
Babies with scalp-ear-nipple syndrome are born with a condition called aplasia cutis congenita, which involves patchy abnormal areas (lesions) on the scalp. These lesions are firm, raised, hairless nodules that resemble open wounds or ulcers at birth, but that heal during childhood.
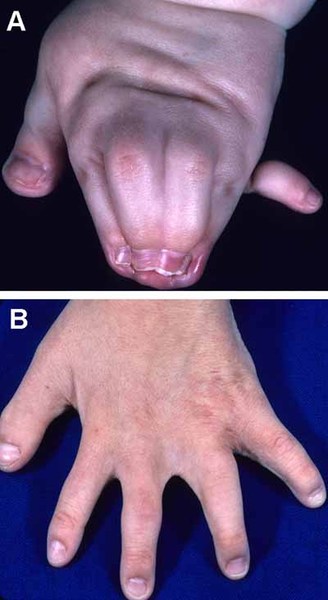
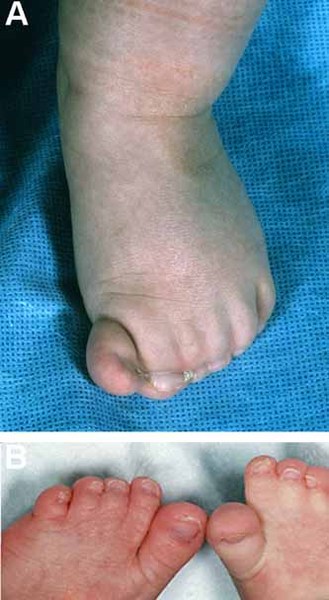
The external ears of people with scalp-ear-nipple syndrome may be small, cup-shaped, folded over, or otherwise mildly misshapen. Hearing is generally normal. Affected individuals also have nipples that are underdeveloped (hypothelia) or absent (athelia). In some cases the underlying breast tissue is absent as well (amastia).
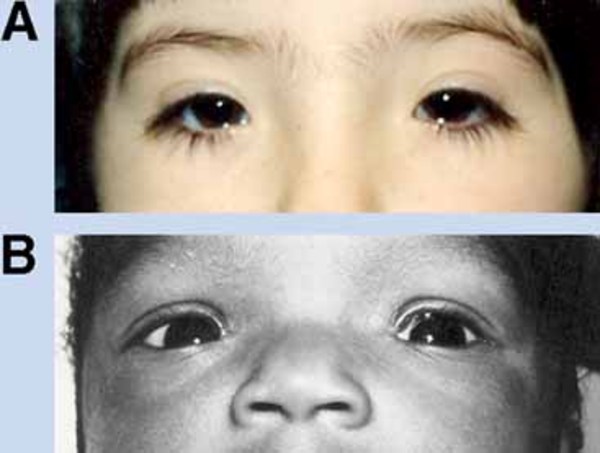
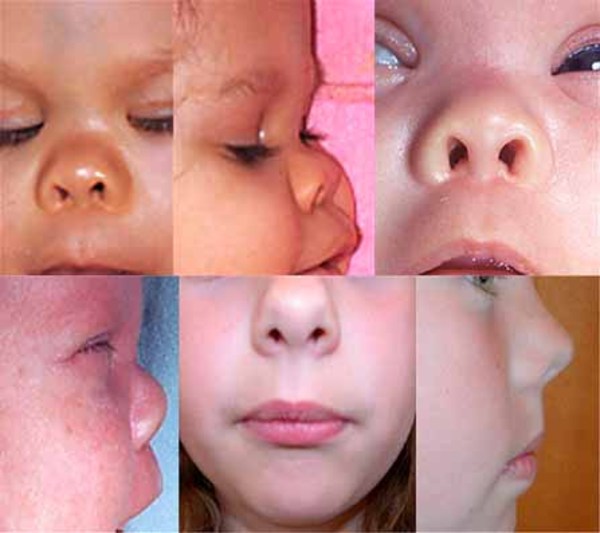
Other features that can occur in this disorder include malformed and brittle fingernails and toenails (nail dystrophy), dental abnormalities including widely-spaced or missing teeth, fusion of the skin between some of the fingers and toes (cutaneous syndactyly), and kidney defects such as underdevelopment (hypoplasia) of one or both kidneys. Unusual facial features, including narrowed openings of the eyes (narrowed palpebral fissures), an increased distance between the inner corners of the eyes (telecanthus), a flat bridge of the nose, and nostrils that open to the front rather than downward (anteverted nares), can also occur in this disorder.
Frequency
The prevalence of scalp-ear-nipple syndrome is unknown. Only a small number of affected individuals have been described in the medical literature.
Causes
Scalp-ear-nipple syndrome is caused by mutations in the KCTD1 gene. This gene provides instructions for making a protein that acts as a transcriptional repressor, which means that it turns off (represses) the activity of certain genes when they are not needed. Specifically, the KCTD1 protein is thought to control (regulate) the activity of genes involved in the development of an embryonic cell layer called the ectoderm. Within the developing embryo, the ectoderm gives rise to several body tissues including the skin, hair, nails, and teeth.
The mutations in the KCTD1 gene that cause scalp-ear-nipple syndrome impair the transcriptional repressor function of the KCTD1 protein. Impairment of this function results in abnormal regulation of genes involved in ectodermal development. The altered gene activity disrupts normal development of the tissues that arise from the ectoderm (ectodermal dysplasia) and leads to the signs and symptoms of scalp-ear-nipple syndrome.
Inheritance
Scalp-ear-nipple syndrome is considered to be an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases of this condition result from new mutations in the gene and occur in people with no history of the disorder in their family. In other cases, an affected person inherits the mutation from one affected parent.


Other Names for This Condition
- Finlay-Marks syndrome
- Hereditary syndrome of lumpy scalp, odd ears, and rudimentary nipples
- SEN syndrome
- SENS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Marneros AG, Beck AE, Turner EH, McMillin MJ, Edwards MJ, Field M, de Macena Sobreira NL, Perez AB, Fortes JA, Lampe AK, Giovannucci Uzielli ML, Gordon CT, Plessis G, Le Merrer M, Amiel J, Reichenberger E, Shively KM, Cerrato F, Labow BI, Tabor HK, Smith JD, Shendure J, Nickerson DA, Bamshad MJ; University of Washington Center for Mendelian Genomics. Mutations in KCTD1 cause scalp-ear-nipple syndrome. Am J Hum Genet. 2013 Apr 4;92(4):621-6. doi: 10.1016/j.ajhg.2013.03.002. Epub 2013 Mar 28. Citation on PubMed or Free article on PubMed Central
- Naik P, Kini P, Chopra D, Gupta Y. Finlay-Marks syndrome: report of two siblings and review of literature. Am J Med Genet A. 2012 Jul;158A(7):1696-701. doi: 10.1002/ajmg.a.35389. Epub 2012 May 25. Citation on PubMed
- Sobreira NL, Brunoni D, Cernach MC, Perez AB. Finlay-Marks (SEN) syndrome: a sporadic case and the delineation of the syndrome. Am J Med Genet A. 2006 Feb 1;140(3):300-2. doi: 10.1002/ajmg.a.31063. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.