

Description
Rhabdoid tumor predisposition syndrome (RTPS) is characterized by a high risk of developing cancerous (malignant) growths called rhabdoid tumors. These highly aggressive tumors are called rhabdoid because their cells resemble rhabdomyoblasts, which are cells that are normally found in embryos before birth and develop into muscles used for movement (skeletal muscles).
Rhabdoid tumors are rare in the general population. They usually occur in the first year of life, and are much less likely to appear after age 4. In people with RTPS, the tumors occur at an average age of 4 to 7 months, and can even occur before birth. Affected individuals may have multifocal synchronous tumors, which means that multiple tumors that develop independently (primary tumors) occur at the same time. The rhabdoid tumors that occur in RTPS usually grow and spread more quickly than those in children without this predisposition, and affected individuals often do not survive past childhood.
More than half of all malignant rhabdoid tumors (MRTs) develop in the cerebellum, which is the part of the brain that coordinates movement. Rhabdoid tumors in the brain and spinal cord (central nervous system) are called atypical teratoid/rhabdoid tumors (AT/RTs).
Rhabdoid tumors also occur outside the central nervous system. These tumors include rhabdoid tumors of the kidneys (RTKs) and tumors that develop in other organs and tissues of the body (called extrarenal malignant rhabdoid tumors or eMRTs). The type of rhabdoid tumor can vary among individuals with RTPS, even within the same family.

Tumors other than rhabdoid tumors can also occur in people with RTPS. Some affected children develop noncancerous (benign) tumors called schwannomas, which grow on nerve cells. Women with RTPS are at increased risk of developing a rare type of ovarian cancer called small cell cancer of the ovary hypercalcemic type (SCCOHT).

Frequency
In the United States, rhabdoid tumors occur in about 1 per million children under age 15. RTPS is thought to account for between a quarter and a third of these tumors.
Causes
In 85 to 95 percent of affected individuals, RTPS is caused by mutations in the SMARCB1 gene. These cases are sometimes known as RTPS1. A small number of cases (called RTPS2) are caused by mutations in the SMARCA4 gene. These genes provide instructions for making proteins that form pieces (subunits) of several different protein groups called SWI/SNF protein complexes. SWI/SNF complexes regulate gene activity (expression) by a process known as chromatin remodeling. Chromatin is the network of DNA and protein that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly DNA is packaged. Chromatin remodeling is one way gene expression is regulated during development; when DNA is tightly packed, gene expression is lower than when DNA is loosely packed.
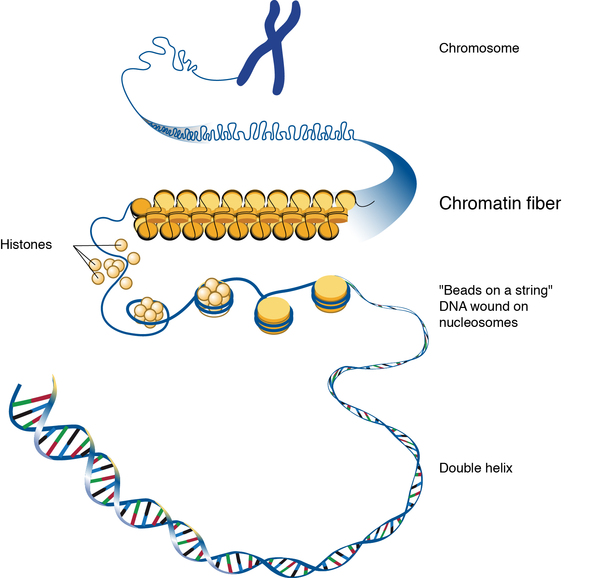
Through their ability to regulate gene activity, SWI/SNF complexes are involved in many processes, including repairing damaged DNA; copying (replicating) DNA; and controlling the growth, division, and maturation (differentiation) of cells. Through these processes, the proteins produced from the SMARCB1 and SMARCA4 genes, as well as other SWI/SNF subunits, are thought to act as tumor suppressors, which keep cells from growing and dividing too rapidly or in an uncontrolled way.
RTPS is caused by a mutation in the SMARCB1 or SMARCA4 gene that is present in cells throughout the body (called a germline mutation). An additional genetic change that deletes the normal copy of the gene is needed for a tumor to develop. This additional change is acquired during a person's lifetime and is present only in the cancerous cells. Such changes are known as somatic mutations. In combination, the germline and somatic mutations lead to the absence or dysfunction of the protein produced from the SMARCB1 or SMARCA4 gene. This deficiency likely impairs the tumor suppressor function of the proteins, but the specific mechanism that leads to rhabdoid tumors is unknown.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to predispose the affected individual to rhabdoid tumors.
The majority of cases of RTPS are caused by SMARCB1 gene mutations. These cases usually occur in people with no history of the disorder in their family. They result from a new mutation in the gene that occurs in a parent's egg or sperm cell and is found in all the child's cells.

In most known cases of RTPS caused by SMARCA4 gene mutations, an affected person inherits the mutation from one parent who has the altered gene but has not developed any rhabdoid tumors.

Other Names for This Condition
- Familial posterior fossa brain tumor of infancy
- Familial posterior fossa brain tumor syndrome
- Familial rhabdoid tumor
- Hereditary SWI/SNF deficiency syndrome
- Rhabdoid predisposition syndrome
- RTPS
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Agaimy A, Foulkes WD. Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol. 2018 May;35(3):193-198. doi: 10.1053/j.semdp.2018.01.002. Epub 2018 Feb 1. Citation on PubMed
- Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011 Jan;56(1):7-15. doi: 10.1002/pbc.22831. Citation on PubMed or Free article on PubMed Central
- Gigante L, Paganini I, Frontali M, Ciabattoni S, Sangiuolo FC, Papi L. Rhabdoid tumor predisposition syndrome caused by SMARCB1 constitutional deletion: prenatal detection of new case of recurrence in siblings due to gonadal mosaicism. Fam Cancer. 2016 Jan;15(1):123-6. doi: 10.1007/s10689-015-9836-6. Citation on PubMed
- Hulsebos TJ, Kenter S, Verhagen WI, Baas F, Flucke U, Wesseling P. Premature termination of SMARCB1 translation may be followed by reinitiation in schwannomatosis-associated schwannomas, but results in absence of SMARCB1 expression in rhabdoid tumors. Acta Neuropathol. 2014 Sep;128(3):439-48. doi: 10.1007/s00401-014-1281-3. Epub 2014 Apr 17. Citation on PubMed
- Kim KH, Roberts CW. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 2014 Sep;207(9):365-72. doi: 10.1016/j.cancergen.2014.04.004. Epub 2014 Apr 13. Citation on PubMed or Free article on PubMed Central
- Kordes U, Bartelheim K, Modena P, Massimino M, Biassoni V, Reinhard H, Hasselblatt M, Schneppenheim R, Fruhwald MC. Favorable outcome of patients affected by rhabdoid tumors due to rhabdoid tumor predisposition syndrome (RTPS). Pediatr Blood Cancer. 2014 May;61(5):919-21. doi: 10.1002/pbc.24793. Epub 2013 Oct 3. Citation on PubMed
- Medina PP, Sanchez-Cespedes M. Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics. 2008 Mar-Apr;3(2):64-8. doi: 10.4161/epi.3.2.6153. Epub 2008 Apr 17. Citation on PubMed
- Schneppenheim R, Fruhwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, Kreuz M, Leuschner I, Martin Subero JI, Obser T, Oyen F, Vater I, Siebert R. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010 Feb 12;86(2):279-84. doi: 10.1016/j.ajhg.2010.01.013. Epub 2010 Feb 4. Citation on PubMed or Free article on PubMed Central
- Sredni ST, Tomita T. Rhabdoid tumor predisposition syndrome. Pediatr Dev Pathol. 2015 Jan-Feb;18(1):49-58. doi: 10.2350/14-07-1531-MISC.1. Epub 2014 Dec 10. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.