

Description
Primary coenzyme Q10 deficiency is a disorder that can affect many parts of the body, especially the brain, muscles, and kidneys. As its name suggests, the disorder involves a shortage (deficiency) of a substance called coenzyme Q10.
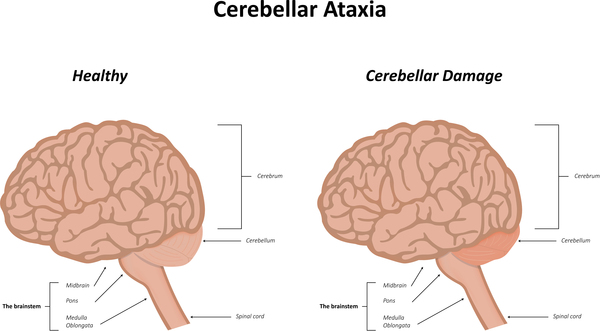
The severity, combination of signs and symptoms, and age of onset of primary coenzyme Q10 deficiency vary widely. In the most severe cases, the condition becomes apparent in infancy and causes severe brain dysfunction combined with muscle weakness (encephalomyopathy) and the failure of other body systems. These problems can be life-threatening. The mildest cases of primary coenzyme Q10 deficiency can begin as late as a person's sixties and often cause cerebellar ataxia, which refers to problems with coordination and balance due to defects in the part of the brain that is involved in coordinating movement (cerebellum). Other neurological abnormalities that can occur in primary coenzyme Q10 deficiency include seizures, intellectual disability, poor muscle tone (hypotonia), involuntary muscle contractions (dystonia), progressive muscle stiffness (spasticity), abnormal eye movements (nystagmus), vision loss caused by degeneration (atrophy) of the optic nerves or breakdown of the light-sensing tissue at the back of the eyes (retinopathy), and sensorineural hearing loss (which is caused by abnormalities in the inner ear). The neurological problems gradually get worse unless treated with coenzyme Q10 supplementation.


A type of kidney dysfunction called nephrotic syndrome is another common feature of primary coenzyme Q10 deficiency. It can occur with or without neurological abnormalities. Nephrotic syndrome occurs when damage to the kidneys impairs their function, which allows protein from the blood to pass into the urine (proteinuria). Other signs and symptoms of nephrotic syndrome include increased cholesterol in the blood (hypercholesterolemia), an abnormal buildup of fluid in the abdominal cavity (ascites), and swelling (edema). Affected individuals may also have blood in the urine (hematuria), which can lead to a reduced number of red blood cells in the body (anemia), abnormal blood clotting, or reduced amounts of certain white blood cells. Low white blood cell counts can lead to a weakened immune system and frequent infections in people with nephrotic syndrome. If not treated with coenzyme Q10 supplementation, affected individuals eventually develop irreversible kidney failure (end-stage renal disease).

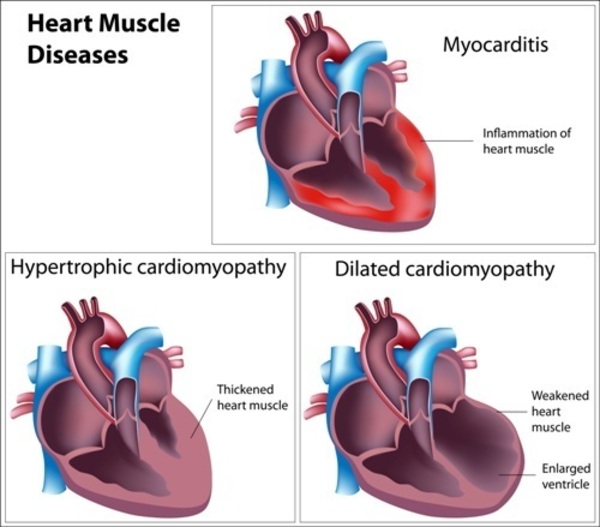
A type of heart disease that enlarges and weakens the heart muscle (hypertrophic cardiomyopathy) can also occur in primary coenzyme Q10 deficiency.
Frequency
The prevalence of primary coenzyme Q10 deficiency is thought to be less than 1 in 100,000 people.
Causes
Primary coenzyme Q10 deficiency is caused by mutations in genes that provide instructions for making proteins involved in the production (synthesis) of a molecule called coenzyme Q10. Collectively, they are called the COQ genes. Most of the identified mutations have occurred in the COQ2, COQ4, COQ6, COQ8A, and COQ8B genes. Smaller numbers of mutations in other COQ genes have also been found to cause primary coenzyme Q10 deficiency.
The coenzyme Q10 molecule has several critical functions in cells throughout the body. In cell structures called mitochondria, coenzyme Q10 plays an essential role in a process called oxidative phosphorylation, which converts the energy from food into a form cells can use. Coenzyme Q10 is also involved in producing pyrimidines, which are building blocks of DNA, its chemical cousin RNA, and molecules such as ATP and GTP that serve as energy sources in the cell. In cell membranes, coenzyme Q10 acts as an antioxidant, protecting cells from damage caused by unstable oxygen-containing molecules (free radicals), which are byproducts of energy production.

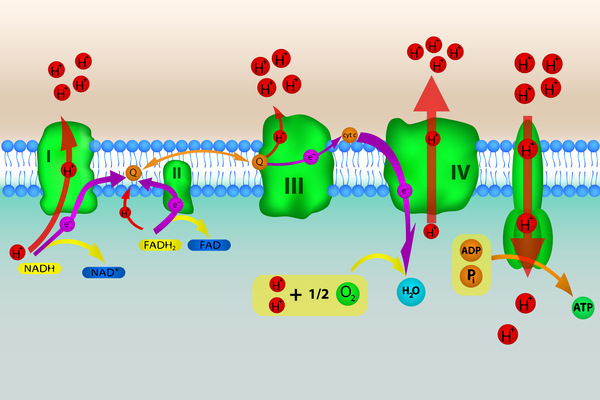
Some mutations in the COQ genes greatly reduce or eliminate the production of the corresponding proteins; others change the structure of a protein, impairing its function. A lack of functional protein produced from any one of the COQ genes decreases the normal production of coenzyme Q10. Studies suggest that a shortage (deficiency) of coenzyme Q10 impairs oxidative phosphorylation and increases the vulnerability of cells to damage from free radicals. A deficiency of coenzyme Q10 may also disrupt the production of pyrimidines. These changes can cause cells throughout the body to malfunction, which may help explain the variety of organs and tissues that can be affected by primary coenzyme Q10 deficiency.
Coenzyme Q10 deficiency can also be caused by mutations in genes that are not directly related to the synthesis of coenzyme Q10. In these cases, the condition is referred to as secondary coenzyme Q10 deficiency. Secondary coenzyme Q10 deficiency is a common feature of certain other genetic conditions.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Coenzyme Q deficiency
- CoQ deficiency
- Primary CoQ10 deficiency
- Ubiquinone deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
- COENZYME Q10 DEFICIENCY, PRIMARY, 1; COQ10D1
- COENZYME Q10 DEFICIENCY, PRIMARY, 2; COQ10D2
- COENZYME Q10 DEFICIENCY, PRIMARY, 3; COQ10D3
- COENZYME Q10 DEFICIENCY, PRIMARY, 4; COQ10D4
- COENZYME Q10 DEFICIENCY, PRIMARY, 5; COQ10D5
- COENZYME Q10 DEFICIENCY, PRIMARY, 6; COQ10D6
- COENZYME Q10 DEFICIENCY, PRIMARY, 7; COQ10D7
- COENZYME Q10 DEFICIENCY, PRIMARY, 8; COQ10D8
- NEPHROTIC SYNDROME, TYPE 9; NPHS9
Scientific Articles on PubMed
References
- Acosta MJ, Vazquez Fonseca L, Desbats MA, Cerqua C, Zordan R, Trevisson E, Salviati L. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta. 2016 Aug;1857(8):1079-1085. doi: 10.1016/j.bbabio.2016.03.036. Epub 2016 Apr 7. Citation on PubMed
- Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J Inherit Metab Dis. 2015 Jan;38(1):145-56. doi: 10.1007/s10545-014-9749-9. Epub 2014 Aug 5. Citation on PubMed
- Doimo M, Desbats MA, Cerqua C, Cassina M, Trevisson E, Salviati L. Genetics of coenzyme q10 deficiency. Mol Syndromol. 2014 Jul;5(3-4):156-62. doi: 10.1159/000362826. Citation on PubMed or Free article on PubMed Central
- Emmanuele V, Lopez LC, Berardo A, Naini A, Tadesse S, Wen B, D'Agostino E, Solomon M, DiMauro S, Quinzii C, Hirano M. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol. 2012 Aug;69(8):978-83. doi: 10.1001/archneurol.2012.206. Citation on PubMed or Free article on PubMed Central
- Laredj LN, Licitra F, Puccio HM. The molecular genetics of coenzyme Q biosynthesis in health and disease. Biochimie. 2014 May;100:78-87. doi: 10.1016/j.biochi.2013.12.006. Epub 2013 Dec 16. Citation on PubMed
- Ozaltin F. Primary coenzyme Q10 (CoQ 10) deficiencies and related nephropathies. Pediatr Nephrol. 2014 Jun;29(6):961-9. doi: 10.1007/s00467-013-2482-z. Epub 2013 Jun 5. Citation on PubMed
- Quinzii CM, Emmanuele V, Hirano M. Clinical presentations of coenzyme q10 deficiency syndrome. Mol Syndromol. 2014 Jul;5(3-4):141-6. doi: 10.1159/000360490. Citation on PubMed or Free article on PubMed Central
- Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Dev Disabil Res Rev. 2010;16(2):183-8. doi: 10.1002/ddrr.108. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.