

Description
Prader-Willi syndrome is a complex genetic condition that affects many parts of the body. In infancy, this condition is characterized by weak muscle tone (hypotonia), feeding difficulties, poor growth, and delayed development. Beginning in childhood, affected individuals develop an extreme hunger, which leads to chronic overeating (hyperphagia) and obesity. Some people with Prader-Willi syndrome, particularly those with obesity, also develop type 2 diabetes (the most common form of diabetes).
People with Prader-Willi syndrome typically have mild to moderate intellectual impairment and learning disabilities. Behavioral problems are common, including temper outbursts, stubbornness, and compulsive behavior such as picking at the skin. Sleep abnormalities can also occur. Additional features of this condition include distinctive facial features such as a narrow forehead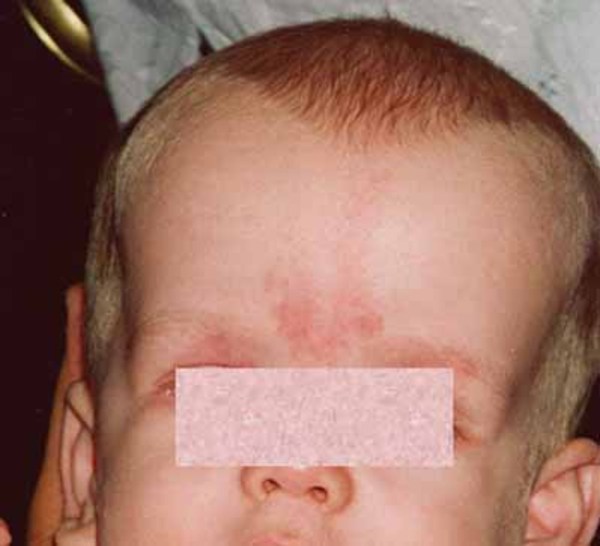 , almond-shaped eyes, and a triangular mouth; short stature; and small hands
, almond-shaped eyes, and a triangular mouth; short stature; and small hands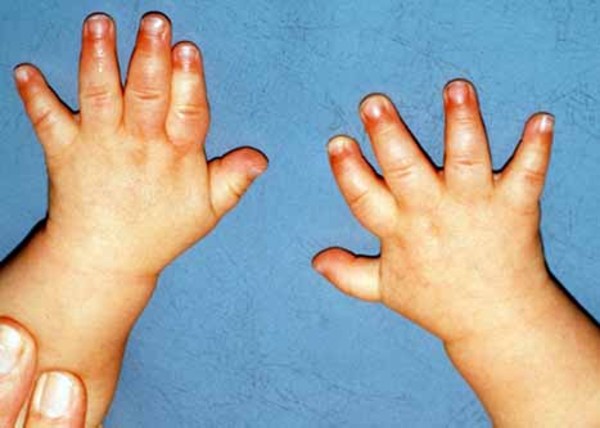 and feet
and feet . Some people with Prader-Willi syndrome have unusually fair skin and light-colored hair
. Some people with Prader-Willi syndrome have unusually fair skin and light-colored hair . Both affected males and affected females have underdeveloped genitals. Puberty is delayed or incomplete, and most affected individuals are unable to have children (infertile).
. Both affected males and affected females have underdeveloped genitals. Puberty is delayed or incomplete, and most affected individuals are unable to have children (infertile).
Frequency
Prader-Willi syndrome affects an estimated 1 in 10,000 to 30,000 people worldwide.
Causes
Prader-Willi syndrome is caused by the loss of function of genes in a particular region of chromosome 15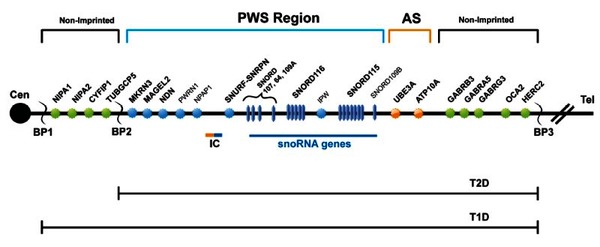 . People normally inherit one copy of this chromosome from each parent. Some genes are turned on (active) only on the copy that is inherited from a person's father (the paternal copy). This parent-specific gene activity results from a process called genomic imprinting.
. People normally inherit one copy of this chromosome from each parent. Some genes are turned on (active) only on the copy that is inherited from a person's father (the paternal copy). This parent-specific gene activity results from a process called genomic imprinting.
Most cases of Prader-Willi syndrome (about 70 percent) occur when a segment of the paternal chromosome 15 is deleted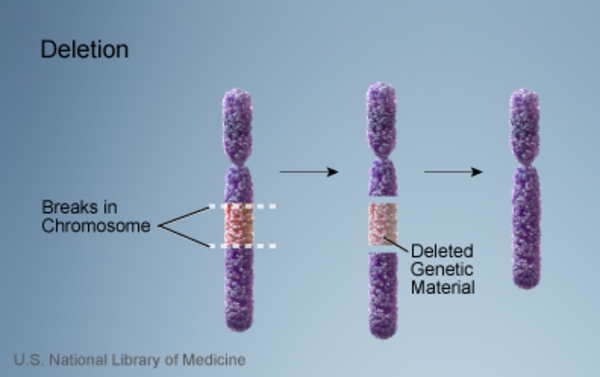 in each cell. People with this chromosomal change are missing certain critical genes in this region because the genes on the paternal copy have been deleted, and the genes on the maternal copy are turned off (inactive). In another 25 percent of cases, a person with Prader-Willi syndrome has two copies of chromosome 15 inherited from his or her mother (maternal copies) instead of one copy from each parent. This situation is called maternal uniparental disomy
in each cell. People with this chromosomal change are missing certain critical genes in this region because the genes on the paternal copy have been deleted, and the genes on the maternal copy are turned off (inactive). In another 25 percent of cases, a person with Prader-Willi syndrome has two copies of chromosome 15 inherited from his or her mother (maternal copies) instead of one copy from each parent. This situation is called maternal uniparental disomy . Rarely, Prader-Willi syndrome can also be caused by a chromosomal rearrangement called a translocation
. Rarely, Prader-Willi syndrome can also be caused by a chromosomal rearrangement called a translocation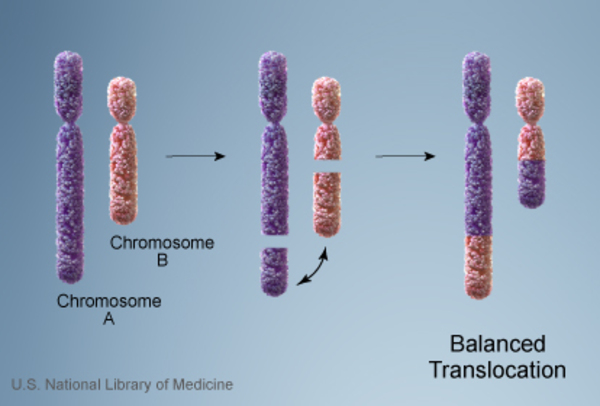 , or by a genetic alteration or other change that abnormally turns off (inactivates) genes on the paternal chromosome 15.
, or by a genetic alteration or other change that abnormally turns off (inactivates) genes on the paternal chromosome 15.
It appears likely that the characteristic features of Prader-Willi syndrome result from the loss of function of several genes on chromosome 15. Among these are genes that provide instructions for making molecules called small nucleolar RNAs (snoRNAs). These molecules have a variety of functions, including helping to regulate other types of RNA molecules. (RNA molecules play essential roles in producing proteins and in other cell activities.) Studies suggest that the loss of a particular group of snoRNA genes, known as the SNORD116 cluster, may play a major role in causing the signs and symptoms of Prader-Willi syndrome. However, it is unknown how a missing SNORD116 cluster could contribute to intellectual disability, behavioral problems, and the physical features of the disorder.
In some people with Prader-Willi syndrome, the loss of a gene called OCA2 is associated with unusually fair skin and light-colored hair. The OCA2 gene is located on the segment of chromosome 15 that is often deleted in people with this disorder. However, loss of the OCA2 gene does not cause the other signs and symptoms of Prader-Willi syndrome. The protein produced from this gene helps determine the coloring (pigmentation) of the skin, hair, and eyes.
Researchers are studying other genes on chromosome 15 that may also be related to the major signs and symptoms of this condition.
Inheritance
Most cases of Prader-Willi syndrome are not inherited, particularly those caused by a deletion in the paternal chromosome 15 or by maternal uniparental disomy. These genetic changes occur as random events during the formation of reproductive cells (eggs and sperm) or in early embryonic development. Affected people typically have no history of the disorder in their family.
Rarely, a genetic change responsible for Prader-Willi syndrome can be inherited. For example, it is possible for a genetic change that abnormally turns off genes on the paternal chromosome 15 to be passed from one generation to the next.
Other Names for This Condition
- Prader-Labhart-Willi syndrome
- PWS
- Willi-Prader syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005 Jul 25;7(14):1-20. doi: 10.1017/S1462399405009531. Citation on PubMed
- Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet. 2009 Jan;17(1):3-13. doi: 10.1038/ejhg.2008.165. Epub 2008 Sep 10. Citation on PubMed or Free article on PubMed Central
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012 Jan;14(1):10-26. doi: 10.1038/gim.0b013e31822bead0. Epub 2011 Sep 26. Citation on PubMed
- Chen C, Visootsak J, Dills S, Graham JM Jr. Prader-Willi syndrome: an update and review for the primary pediatrician. Clin Pediatr (Phila). 2007 Sep;46(7):580-91. doi: 10.1177/0009922807299314. Epub 2007 May 23. Citation on PubMed
- Driscoll DJ, Miller JL, Cassidy SB. Prader-Willi Syndrome. 1998 Oct 6 [updated 2026 Feb 19]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1330/ Citation on PubMed
- Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M; speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab. 2008 Nov;93(11):4183-97. doi: 10.1210/jc.2008-0649. Epub 2008 Aug 12. Citation on PubMed
- Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001 Nov;108(5):E92. doi: 10.1542/peds.108.5.e92. Citation on PubMed
- Kocher MA, Huang FW, Le E, Good DJ. Snord116 Post-transcriptionally Increases Nhlh2 mRNA Stability: Implications for Human Prader-Willi Syndrome. Hum Mol Genet. 2021 Jun 9;30(12):1101-1110. doi: 10.1093/hmg/ddab103. Citation on PubMed
- Lee S, Wevrick R. Identification of novel imprinted transcripts in the Prader-Willi syndrome and Angelman syndrome deletion region: further evidence for regional imprinting control. Am J Hum Genet. 2000 Mar;66(3):848-58. doi: 10.1086/302817. Citation on PubMed or Free article on PubMed Central
- Mendiola AJP, LaSalle JM. Epigenetics in Prader-Willi Syndrome. Front Genet. 2021 Feb 15;12:624581. doi: 10.3389/fgene.2021.624581. eCollection 2021. Citation on PubMed
- Oiglane-Shlik E, Zordania R, Varendi H, Antson A, Magi ML, Tasa G, Bartsch O, Talvik T, Ounap K. The neonatal phenotype of Prader-Willi syndrome. Am J Med Genet A. 2006 Jun 1;140(11):1241-4. doi: 10.1002/ajmg.a.31223. No abstract available. Citation on PubMed
- Shelkowitz E, Gantz MG, Ridenour TA, Scheimann AO, Strong T, Bohonowych J, Duis J. Neuropsychiatric features of Prader-Willi syndrome. Am J Med Genet A. 2022 May;188(5):1457-1463. doi: 10.1002/ajmg.a.62662. Epub 2022 Jan 30. Citation on PubMed
- Wattendorf DJ, Muenke M. Prader-Willi syndrome. Am Fam Physician. 2005 Sep 1;72(5):827-30. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.