

Description
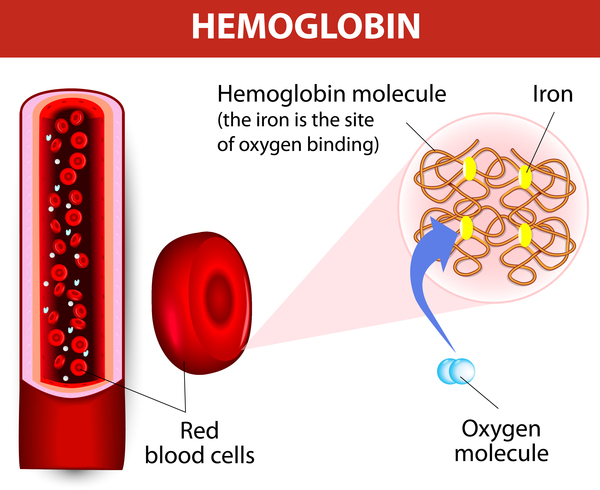
Porphyria is a group of disorders caused by abnormalities in the chemical steps that lead to heme production. Heme is a vital molecule for all of the body's organs, although it is most abundant in the blood, bone marrow, and liver. Heme is a component of several iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
Researchers have identified several types of porphyria, which are distinguished by their genetic cause and their signs and symptoms. Some types of porphyria, called cutaneous porphyrias, primarily affect the skin. Areas of skin exposed to the sun become fragile and blistered, which can lead to infection, scarring, changes in skin coloring (pigmentation), and increased hair growth. Cutaneous porphyrias include congenital erythropoietic porphyria, erythropoietic protoporphyria, hepatoerythropoietic porphyria, and porphyria cutanea tarda.
Other types of porphyria, called acute porphyrias, primarily affect the nervous system. These disorders are described as "acute" because their signs and symptoms appear quickly and usually last a short time. Episodes of acute porphyria can cause abdominal pain, vomiting, constipation, and diarrhea. During an episode, a person may also experience muscle weakness, seizures, fever, and mental changes such as anxiety and hallucinations. These signs and symptoms can be life-threatening, especially if the muscles that control breathing become paralyzed. Acute porphyrias include acute intermittent porphyria and ALAD deficiency porphyria. Two other forms of porphyria, hereditary coproporphyria and variegate porphyria, can have both acute and cutaneous symptoms.
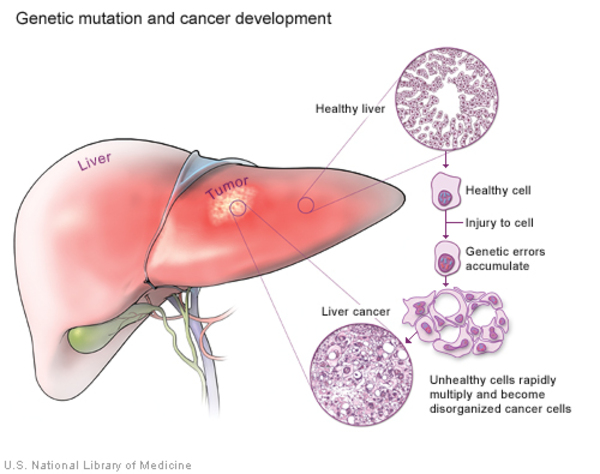
The porphyrias can also be split into erythropoietic and hepatic types, depending on where damaging compounds called porphyrins and porphyrin precursors first build up in the body. In erythropoietic porphyrias, these compounds originate in the bone marrow. Erythropoietic porphyrias include erythropoietic protoporphyria and congenital erythropoietic porphyria. Health problems associated with erythropoietic porphyrias include a low number of red blood cells (anemia) and enlargement of the spleen (splenomegaly). The other types of porphyrias are considered hepatic porphyrias. In these disorders, porphyrins and porphyrin precursors originate primarily in the liver, leading to abnormal liver function and an increased risk of developing liver cancer.


Environmental factors can strongly influence the occurrence and severity of signs and symptoms of porphyria. Alcohol, smoking, certain drugs, hormones, other illnesses, stress, and dieting or periods without food (fasting) can all trigger the signs and symptoms of some forms of the disorder. Additionally, exposure to sunlight worsens the skin damage in people with cutaneous porphyrias.
Frequency
The exact prevalence of porphyria is unknown, but it probably ranges from 1 in 500 to 1 in 50,000 people worldwide. Overall, porphyria cutanea tarda is the most common type of porphyria. For some forms of porphyria, the prevalence is unknown because many people with a genetic mutation associated with the disease never experience signs or symptoms.
Acute intermittent porphyria is the most common form of acute porphyria in most countries. It may occur more frequently in northern European countries, such as Sweden, and in the United Kingdom. Another form of the disorder, hereditary coproporphyria, has been reported mostly in Europe and North America. Variegate porphyria is most common in the Afrikaner population of South Africa; about 3 in 1,000 people in this population have the genetic change that causes this form of the disorder.
Causes
Each form of porphyria results from mutations in one of these genes: ALAD, ALAS2, CPOX, FECH, HMBS, PPOX, UROD, or UROS.
The genes related to porphyria provide instructions for making the enzymes needed to produce heme. Mutations in most of these genes reduce enzyme activity, which limits the amount of heme the body can produce. As a result, compounds called porphyrins and porphyrin precursors, which are formed during the process of heme production, can build up abnormally in the liver and other organs. When these substances accumulate in the skin and interact with sunlight, they cause the cutaneous forms of porphyria. The acute forms of the disease occur when porphyrins and porphyrin precursors build up in and damage the nervous system.
One type of porphyria, porphyria cutanea tarda, results from both genetic and nongenetic factors. About 20 percent of cases are related to mutations in the UROD gene. The remaining cases are not associated with UROD gene mutations and are classified as sporadic. Many factors contribute to the development of porphyria cutanea tarda. These include an increased amount of iron in the liver, alcohol consumption, smoking, hepatitis C or HIV infection, or certain hormones. Mutations in the HFE gene (which cause an iron overload disorder called hemochromatosis) are also associated with porphyria cutanea tarda. Other, as-yet-unidentified genetic factors may also play a role in this form of porphyria.
Inheritance
Some types of porphyria are inherited in an autosomal dominant pattern, which means one copy of the gene in each cell is mutated. This single mutation is sufficient to reduce the activity of an enzyme needed for heme production, which increases the risk of developing signs and symptoms of porphyria. Autosomal dominant porphyrias include acute intermittent porphyria, most cases of erythropoietic protoporphyria, hereditary coproporphyria, and variegate porphyria. Although the gene mutations associated with some cases of porphyria cutanea tarda also have an autosomal dominant inheritance pattern, most people with this form of porphyria do not have an inherited gene mutation.

Other porphyrias are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition. Porphyrias with an autosomal recessive pattern of inheritance include ALAD deficiency porphyria, congenital erythropoietic porphyria, and some cases of erythropoietic protoporphyria.

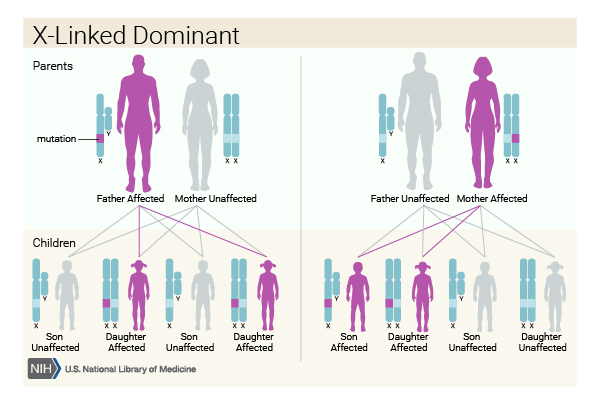
When erythropoietic protoporphyria is caused by mutations in the ALAS2 gene, it has an X-linked dominant pattern of inheritance. The ALAS2 gene is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell may be sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. Males may experience more severe symptoms of the disorder than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

Mutations in the UROD gene are related to both porphyria cutanea tarda and hepatoerythropoietic porphyria. Individuals who inherit one altered copy of the UROD gene are at increased risk for porphyria cutanea tarda. (Multiple genetic and nongenetic factors contribute to this condition.) People who inherit two altered copies of the UROD gene in each cell develop hepatoerythropoietic porphyria.
Other Names for This Condition
- Hematoporphyria
- Porphyrin disorder
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Acute intermittent porphyria

- Genetic Testing Registry: Familial porphyria cutanea tarda
- Genetic Testing Registry: Cutaneous porphyria
- Genetic Testing Registry: X-linked erythropoietic protoporphyria
- Genetic Testing Registry: Hereditary coproporphyria
- Genetic Testing Registry: Porphyria
- Genetic Testing Registry: Protoporphyria, erythropoietic, 1
- Genetic Testing Registry: Variegate porphyria
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005 Mar 15;142(6):439-50. doi: 10.7326/0003-4819-142-6-200503150-00010. Citation on PubMed
- Badminton MN, Elder GH. Molecular mechanisms of dominant expression in porphyria. J Inherit Metab Dis. 2005;28(3):277-86. doi: 10.1007/s10545-005-8050-3. Citation on PubMed
- Chemmanur AT, Bonkovsky HL. Hepatic porphyrias: diagnosis and management. Clin Liver Dis. 2004 Nov;8(4):807-38, viii. doi: 10.1016/j.cld.2004.07.001. Citation on PubMed
- Dombeck TA, Satonik RC. The porphyrias. Emerg Med Clin North Am. 2005 Aug;23(3):885-99, x. doi: 10.1016/j.emc.2005.03.014. Citation on PubMed
- Kauppinen R. Porphyrias. Lancet. 2005 Jan 15-21;365(9455):241-52. doi: 10.1016/S0140-6736(05)17744-7. Citation on PubMed
- Lecha M, Herrero C, Ozalla D. Diagnosis and treatment of the hepatic porphyrias. Dermatol Ther. 2003;16(1):65-72. doi: 10.1046/j.1529-8019.2003.01610.x. Citation on PubMed
- Murphy GM. Diagnosis and management of the erythropoietic porphyrias. Dermatol Ther. 2003;16(1):57-64. doi: 10.1046/j.1529-8019.2003.01609.x. Citation on PubMed
- Nordmann Y, Puy H. Human hereditary hepatic porphyrias. Clin Chim Acta. 2002 Nov;325(1-2):17-37. doi: 10.1016/s0009-8981(02)00276-0. Citation on PubMed
- Peters TJ, Sarkany R. Porphyria for the general physician. Clin Med (Lond). 2005 May-Jun;5(3):275-81. doi: 10.7861/clinmedicine.5-3-275. No abstract available. Citation on PubMed
- Sarkany RP. Making sense of the porphyrias. Photodermatol Photoimmunol Photomed. 2008 Apr;24(2):102-8. doi: 10.1111/j.1600-0781.2008.00336.x. Citation on PubMed
- Sassa S. The porphyrias. Photodermatol Photoimmunol Photomed. 2002 Apr;18(2):56-67. doi: 10.1034/j.1600-0781.2002.180202.x. No abstract available. Citation on PubMed
- Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008 Sep;83(3):408-14. doi: 10.1016/j.ajhg.2008.08.003. Epub 2008 Sep 4. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.