

Description
PACS1 syndrome is a condition in which all affected individuals have intellectual disability, speech and language problems, and a distinct facial appearance. Many affected individuals have additional neurological, behavioral, and health problems.
In PACS1 syndrome, intellectual disability typically ranges from mild to moderate. Individuals with this condition also have problems with producing speech (expressive language). Speech development ranges from limited language to few words or no speech.
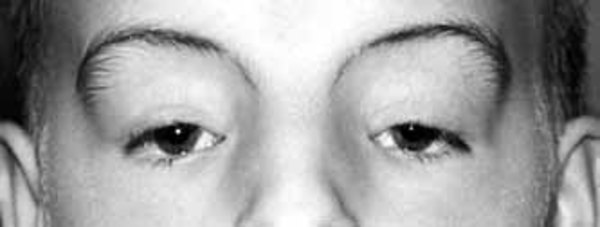
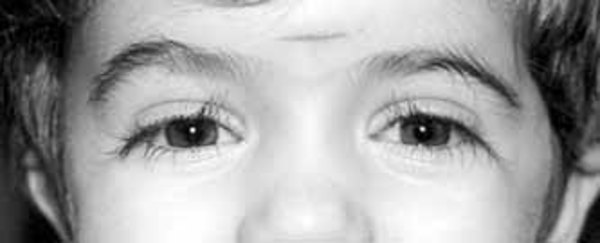
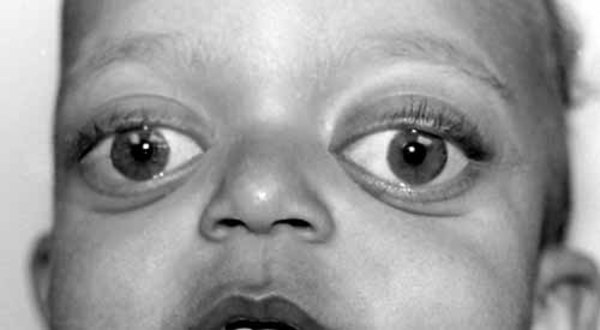
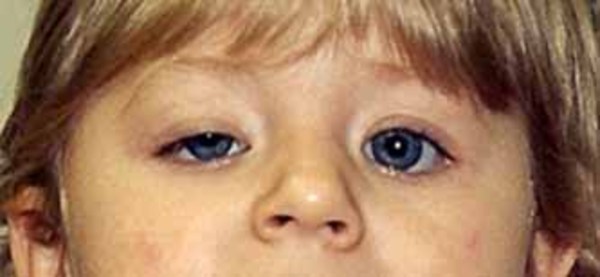
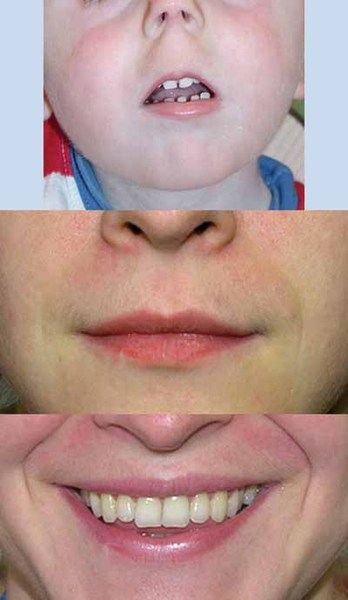
Individuals with PACS1 syndrome have a distinct facial appearance. Facial features include thick and highly arched eyebrows, long eyelashes, widely set eyes (hypertelorism), outside corners of the eyes that point downward (downslanting palpebral fissures), droopy eyelids (ptosis), a rounded nasal tip, a wide mouth with corners that point downward, a thin upper lip, a smooth area between the nose and upper lip (philtrum), widely spaced teeth, and ears that are low-set with fewer folds and grooves than normal (described as "simple"). Abnormalities of other body systems can also occur, such as malformations of the heart, brain, eyes, or other organs. Males may have undescended testes (cryptorchidism).



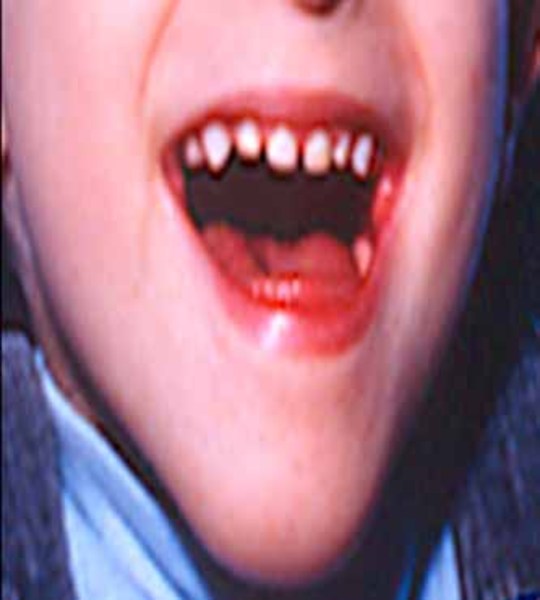
Children with PACS1 syndrome often have problems learning to eat solid food and prefer soft foods. When given solid foods, affected children often swallow without chewing. These food issues tend to persist throughout life. Some affected individuals experience a backflow of stomach acids into the esophagus (gastroesophageal reflux).
Additional neurological problems can occur in PACS1 syndrome. Some affected individuals have features of autism spectrum disorder, which is characterized by impaired communication and social interaction. Attention-deficit/hyperactivity disorder (ADHD), obsessive-compulsive disorder (OCD), self-injury, or frustration leading to tantrums can also occur. Most individuals with PACS1 syndrome have seizures that vary in type and age of onset. Some people with PACS1 syndrome have weak muscle tone (hypotonia). Individuals with this condition are often delayed in walking, with some developing an unsteady walking style (gait). Rarely, affected individuals have frequent falls and gradually lose their ability to walk in late childhood, requiring wheelchair assistance.
Frequency
The prevalence of PACS1 syndrome is unknown; more than 30 affected individuals have been described in the scientific literature.
Causes
PACS1 syndrome is caused by mutations in a gene called PACS1. This gene provides instructions for making a protein that helps transport molecules and other proteins to cells and tissues where they are needed. The PACS1 protein is found in a complex network of membranes known as the trans-Golgi network, which sorts proteins and other molecules and sends them to their intended destinations inside or outside the cell. The PACS1 protein is most active during development before birth.
Almost all cases of PACS1 syndrome are caused by the same mutation. This and other PACS1 gene mutations are thought to impair the protein's ability to aid in the transport of certain molecules and proteins. Such an impairment likely results in the accumulation or misplacement of molecules or proteins within cells; however, the effects of these accumulated substances is unclear. Research suggests that impaired PACS1 protein function disrupts normal development of structures in the face, leading to a distinct facial appearance. It is likely that the development of other body systems are similarly affected by impaired PACS1 protein function, leading to other signs and symptoms of PACS1 syndrome, but more research is needed to understand the mechanisms.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Most cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual’s parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- Autosomal dominant intellectual disability-17
- Intellectual disability-craniofacial dysmorphism-cryptorchidism syndrome
- PACS1-related syndrome
- Schuurs-Hoeijmakers syndrome
- SHMS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dutta AK. Schuurs-Hoeijmakers syndrome in a patient from India. Am J Med Genet A. 2019 Apr;179(4):522-524. doi: 10.1002/ajmg.a.61058. Epub 2019 Jan 28. Citation on PubMed
- Gadzicki D, Docker D, Schubach M, Menzel M, Schmorl B, Stellmer F, Biskup S, Bartholdi D. Expanding the phenotype of a recurrent de novo variant in PACS1 causing intellectual disability. Clin Genet. 2015 Sep;88(3):300-2. doi: 10.1111/cge.12544. Epub 2014 Dec 18. No abstract available. Citation on PubMed
- Hoshino Y, Enokizono T, Imagawa K, Tanaka R, Suzuki H, Fukushima H, Arai J, Sumazaki R, Uehara T, Takenouchi T, Kosaki K. Schuurs-Hoeijmakers syndrome in two patients from Japan. Am J Med Genet A. 2019 Mar;179(3):341-343. doi: 10.1002/ajmg.a.9. Epub 2018 Dec 27. Citation on PubMed
- Lusk L, Smith S, Martin C, Taylor C, Chung W. PACS1 Neurodevelopmental Disorder. 2020 Jul 16. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK559434/ Citation on PubMed
- Martinez-Monseny A, Bolasell M, Arjona C, Martorell L, Yubero D, Arsmtrong J, Maynou J, Fernandez G, Del Carmen Salgado M, Palau F, Serrano M. Mutation of PACS1: the milder end of the spectrum. Clin Dysmorphol. 2018 Oct;27(4):148-150. doi: 10.1097/MCD.0000000000000237. No abstract available. Citation on PubMed
- Miyake N, Ozasa S, Mabe H, Kimura S, Shiina M, Imagawa E, Miyatake S, Nakashima M, Mizuguchi T, Takata A, Ogata K, Matsumoto N. A novel missense mutation affecting the same amino acid as the recurrent PACS1 mutation in Schuurs-Hoeijmakers syndrome. Clin Genet. 2018 Apr;93(4):929-930. doi: 10.1111/cge.13105. Epub 2017 Oct 4. Citation on PubMed
- Pefkianaki M, Schneider A, Capasso JE, Wasserman BN, Bardakjian T, Levin AV. Ocular manifestations of PACS1 mutation. J AAPOS. 2018 Aug;22(4):323-325. doi: 10.1016/j.jaapos.2017.12.008. Epub 2018 Mar 14. Citation on PubMed
- Schuurs-Hoeijmakers JH, Landsverk ML, Foulds N, Kukolich MK, Gavrilova RH, Greville-Heygate S, Hanson-Kahn A, Bernstein JA, Glass J, Chitayat D, Burrow TA, Husami A, Collins K, Wusik K, van der Aa N, Kooy F, Brown KT, Gadzicki D, Kini U, Alvarez S, Fernandez-Jaen A, McGehee F, Selby K, Tarailo-Graovac M, Van Allen M, van Karnebeek CD, Stavropoulos DJ, Marshall CR, Merico D, Gregor A, Zweier C, Hopkin RJ, Chu YW, Chung BH, de Vries BB, Devriendt K, Hurles ME, Brunner HG; DDD study. Clinical delineation of the PACS1-related syndrome--Report on 19 patients. Am J Med Genet A. 2016 Mar;170(3):670-5. doi: 10.1002/ajmg.a.37476. Epub 2016 Feb 3. Citation on PubMed
- Schuurs-Hoeijmakers JH, Oh EC, Vissers LE, Swinkels ME, Gilissen C, Willemsen MA, Holvoet M, Steehouwer M, Veltman JA, de Vries BB, van Bokhoven H, de Brouwer AP, Katsanis N, Devriendt K, Brunner HG. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am J Hum Genet. 2012 Dec 7;91(6):1122-7. doi: 10.1016/j.ajhg.2012.10.013. Epub 2012 Nov 15. Citation on PubMed or Free article on PubMed Central
- Stern D, Cho MT, Chikarmane R, Willaert R, Retterer K, Kendall F, Deardorff M, Hopkins S, Bedoukian E, Slavotinek A, Schrier Vergano S, Spangler B, McDonald M, McConkie-Rosell A, Burton BK, Kim KH, Oundjian N, Kronn D, Chandy N, Baskin B, Guillen Sacoto MJ, Wentzensen IM, McLaughlin HM, McKnight D, Chung WK. Association of the missense variant p.Arg203Trp in PACS1 as a cause of intellectual disability and seizures. Clin Genet. 2017 Aug;92(2):221-223. doi: 10.1111/cge.12956. Epub 2017 Jan 23. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.