

Description
Ornithine translocase deficiency is an inherited disorder that causes ammonia and other substances to build up (accumulate) in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.
Ornithine translocase deficiency varies widely in its severity and age of onset. Affected infants show signs and symptoms of ornithine translocase deficiency within days after birth. In most affected individuals, however, signs and symptoms of ornithine translocase deficiency do not appear until later in life, with health problems first appearing anytime from childhood to adulthood. Later-onset forms of ornithine translocase deficiency are usually less severe than the infantile form.
Infants with ornithine translocase deficiency may lack energy (be lethargic), refuse to eat, vomit frequently, or have poorly controlled breathing or body temperature. Seizures or unusual body movements are common in these individuals. Some people with this condition have intellectual disability or developmental delay, but others have normal intelligence. Severe cases may result in coma.
Some people with later-onset ornithine translocase deficiency have episodes of vomiting, lethargy, problems with coordination (ataxia), vision problems, episodes of brain dysfunction (encephalopathy), developmental delay, learning disabilities, or stiffness caused by abnormal tensing of the muscles (spasticity). Affected individuals may have chronic liver problems and mild abnormal bleeding.
Individuals with ornithine translocase deficiency often cannot tolerate high-protein foods, such as meat. Occasionally, high-protein meals or stress caused by illness or periods without food (fasting) may cause ammonia to accumulate more quickly in the blood. This rapid increase of ammonia likely leads to the signs and symptoms of ornithine translocase deficiency.
While the signs and symptoms of ornithine translocase deficiency can vary greatly among affected individuals, proper treatment can prevent some complications from occurring and may improve quality of life.
Frequency
Ornithine translocase deficiency is a very rare disorder. More than 100 affected individuals have been described in the scientific literature.
Causes
Mutations in the SLC25A15 gene cause ornithine translocase deficiency. The SLC25A15 gene provides instructions for making a protein called mitochondrial ornithine transporter 1. This protein participates in the urea cycle, which is a sequence of biochemical reactions that occurs in liver cells. The urea cycle breaks down excess nitrogen, made when protein is broken down by the body, to make a compound called urea that is excreted by the kidneys in urine.
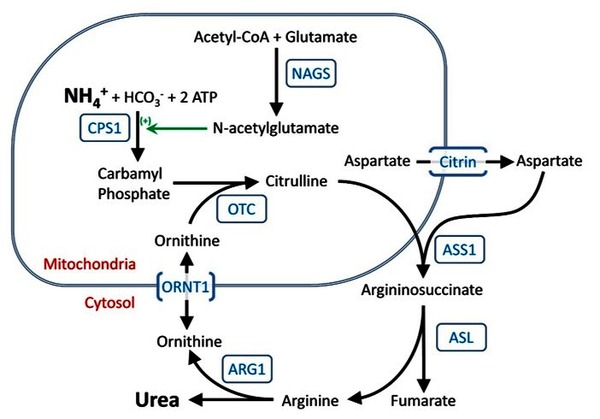

Mitochondrial ornithine transporter 1 is located within the mitochondria (the energy-producing centers in cells), where the protein transports a molecule called ornithine so it can participate in the urea cycle.

Mutations in the SLC25A15 gene cause the production of a mitochondrial ornithine transporter 1 with reduced or absent function. As a result, ornithine transport is impaired and the urea cycle cannot proceed normally. This causes, nitrogen to accumulate in the bloodstream in the form of toxic ammonia instead of being converted to less toxic urea and being excreted. Ammonia is especially damaging to the brain, and excess ammonia causes neurological problems and other signs and symptoms of ornithine translocase deficiency. Byproducts of impaired ornithine transport in people with this condition include the accumulation of a substance called ornithine in the blood (hyperornithinemia) and the excretion of a substance called homocitrulline in the urine (homocitrullinuria).
Another version of the mitochondrial ornithine transporter protein is produced by a different gene. While this protein is not as abundant as mitochondrial ornithine transporter 1, it is thought that this other version of the protein may partially compensate for the loss of mitochondrial ornithine transporter 1 and contribute to the late age of onset and mild signs and symptoms in some affected individuals. Other factors, many unknown, also contribute to the variable severity of ornithine translocase deficiency.
Because ornithine translocase deficiency is caused by problems with the urea cycle, it belongs to a class of genetic diseases called urea cycle disorders.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- HHH syndrome
- Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome
- Hyperornithinemia-hyperammonemia-homocitrullinemia syndrome
- Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome
- Triple H syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Camacho JA, Mardach R, Rioseco-Camacho N, Ruiz-Pesini E, Derbeneva O, Andrade D, Zaldivar F, Qu Y, Cederbaum SD. Clinical and functional characterization of a human ORNT1 mutation (T32R) in the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr Res. 2006 Oct;60(4):423-9. doi: 10.1203/01.pdr.0000238301.25938.f5. Epub 2006 Aug 28. Citation on PubMed
- Camacho JA, Obie C, Biery B, Goodman BK, Hu CA, Almashanu S, Steel G, Casey R, Lambert M, Mitchell GA, Valle D. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet. 1999 Jun;22(2):151-8. doi: 10.1038/9658. Citation on PubMed
- Camacho JA, Rioseco-Camacho N, Andrade D, Porter J, Kong J. Cloning and characterization of human ORNT2: a second mitochondrial ornithine transporter that can rescue a defective ORNT1 in patients with the hyperornithinemia-hyperammonemia-homocitrullinuria syndrome, a urea cycle disorder. Mol Genet Metab. 2003 Aug;79(4):257-71. doi: 10.1016/s1096-7192(03)00105-7. Citation on PubMed
- Haberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012 May 29;7:32. doi: 10.1186/1750-1172-7-32. Citation on PubMed or Free article on PubMed Central
- Haberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Lindner M, Mandel H, Martinelli D, Pintos-Morell G, Santer R, Skouma A, Servais A, Tal G, Rubio V, Huemer M, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J Inherit Metab Dis. 2019 Nov;42(6):1192-1230. doi: 10.1002/jimd.12100. Epub 2019 May 15. Citation on PubMed
- Martinelli D, Diodato D, Ponzi E, Monne M, Boenzi S, Bertini E, Fiermonte G, Dionisi-Vici C. The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Orphanet J Rare Dis. 2015 Mar 11;10:29. doi: 10.1186/s13023-015-0242-9. Citation on PubMed or Free article on PubMed Central
- Miyamoto T, Kanazawa N, Kato S, Kawakami M, Inoue Y, Kuhara T, Inoue T, Takeshita K, Tsujino S. Diagnosis of Japanese patients with HHH syndrome by molecular genetic analysis: a common mutation, R179X. J Hum Genet. 2001;46(5):260-2. doi: 10.1007/s100380170075. Citation on PubMed
- Waisbren SE, Gropman AL; Members of the Urea Cycle Disorders Consortium (UCDC); Batshaw ML. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. J Inherit Metab Dis. 2016 Jul;39(4):573-84. doi: 10.1007/s10545-016-9942-0. Epub 2016 May 23. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.