

Description
Ornithine transcarbamylase deficiency is an inherited disorder that causes ammonia to accumulate in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.
Ornithine transcarbamylase deficiency can become evident at any age. The most severe form occurs in the first few days of life. This neonatal-onset form of the disorder usually affects males; it is very rare in females. An infant with the neonatal-onset form of ornithine transcarbamylase deficiency may be lacking in energy (lethargic) or unwilling to eat, and have a poorly-controlled breathing rate or body temperature. Infants with this disorder may be described as "floppy" and can experience seizures or coma. Complications from ornithine transcarbamylase deficiency may include developmental delay and intellectual disability. Progressive liver damage may also occur.

In some affected individuals, signs and symptoms of ornithine transcarbamylase deficiency may be less severe, and may not appear until later in life. The late-onset form of the disorder occurs in both males and females. People with late-onset ornithine transcarbamylase deficiency may experience episodes of altered mental status, such as delirium, erratic behavior, or a reduced level of consciousness. Headaches, vomiting, aversion to protein foods, and seizures can also occur in this form of the disorder.
Frequency
Estimates of the prevalence of ornithine transcarbamylase deficiency have ranged from 1 in 14,000 to 1 in 77,000 people. Individuals with the neonatal-onset form of the disorder are more likely to be counted in these estimates, because people with the late-onset form are less likely to come to medical attention.
Causes
Mutations in the OTC gene cause ornithine transcarbamylase deficiency. The OTC gene provides instructions for making the ornithine transcarbamylase enzyme.
Ornithine transcarbamylase deficiency belongs to a class of genetic diseases called urea cycle disorders. The urea cycle is a sequence of reactions that occurs in liver cells. It processes excess nitrogen, generated when protein is used by the body, to make a compound called urea that is excreted by the kidneys. The ornithine transcarbamylase enzyme starts a specific reaction within the urea cycle.

In ornithine transcarbamylase deficiency, as its name suggests, the ornithine transcarbamylase enzyme is damaged or missing. The urea cycle cannot proceed normally, and nitrogen accumulates in the bloodstream in the form of ammonia.
Ammonia is especially damaging to the nervous system, so ornithine transcarbamylase deficiency causes neurological problems as well as eventual damage to the liver.
Inheritance
Ornithine transcarbamylase deficiency is an X-linked disorder. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes . A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), mutations in both copies of the gene will cause the disorder. Some females with only one altered copy of the OTC gene also show signs and symptoms of ornithine transcarbamylase deficiency.
In females, who have two copies of the X chromosome, one altered copy of the OTC
gene in each cell can lead to less severe features of the condition or may cause no signs or symptoms at all. However, many females with one altered copy of this gene have ornithine transcarbamylase deficiency similar to affected males because the X chromosome with the normal copy of the OTC gene is turned off through a process called
X-inactivation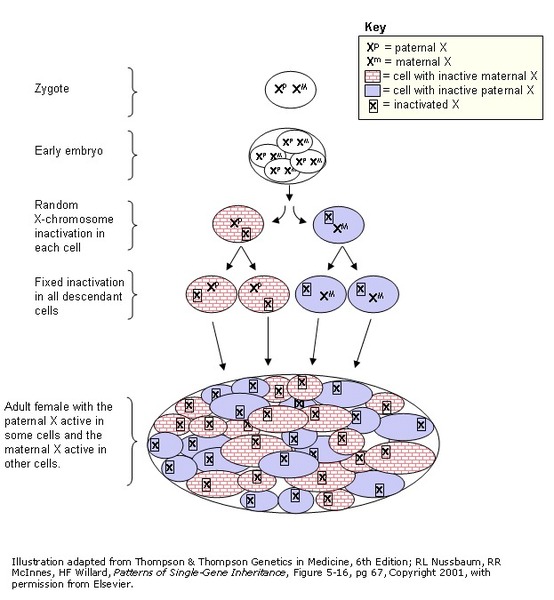 .
Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
.
Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
Other Names for This Condition
- Ornithine Carbamoyltransferase Deficiency Disease
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Brassier A, Gobin S, Arnoux JB, Valayannopoulos V, Habarou F, Kossorotoff M, Servais A, Barbier V, Dubois S, Touati G, Barouki R, Lesage F, Dupic L, Bonnefont JP, Ottolenghi C, De Lonlay P. Long-term outcomes in Ornithine Transcarbamylase deficiency: a series of 90 patients. Orphanet J Rare Dis. 2015 May 10;10:58. doi: 10.1186/s13023-015-0266-1. Citation on PubMed or Free article on PubMed Central
- Caldovic L, Abdikarim I, Narain S, Tuchman M, Morizono H. Genotype-Phenotype Correlations in Ornithine Transcarbamylase Deficiency: A Mutation Update. J Genet Genomics. 2015 May 20;42(5):181-94. doi: 10.1016/j.jgg.2015.04.003. Epub 2015 May 19. Citation on PubMed or Free article on PubMed Central
- Choi JH, Lee BH, Kim JH, Kim GH, Kim YM, Cho J, Cheon CK, Ko JM, Lee JH, Yoo HW. Clinical outcomes and the mutation spectrum of the OTC gene in patients with ornithine transcarbamylase deficiency. J Hum Genet. 2015 Sep;60(9):501-7. doi: 10.1038/jhg.2015.54. Citation on PubMed
- Helman G, Pacheco-Colon I, Gropman AL. The urea cycle disorders. Semin Neurol. 2014 Jul;34(3):341-9. doi: 10.1055/s-0034-1386771. Epub 2014 Sep 5. Citation on PubMed
- Simpson KL, MacLeod EL, Kakajiwala A, Gropman AL, Ah Mew N. Urea Cycle Disorders Overview. 2003 Apr 29 [updated 2025 Jul 3]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1217/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.