

Description
Optic atrophy type 1 is a condition that often causes slowly worsening vision, usually beginning in childhood. People with optic atrophy type 1 typically experience a narrowing of their field of vision (tunnel vision). Affected individuals gradually lose their sight as their field of vision becomes smaller. Both eyes are usually affected equally, but the severity of the vision loss varies widely, even among affected members of the same family, ranging from nearly normal vision to complete blindness.
In addition to vision loss, people with optic atrophy type 1 frequently have problems with color vision (color vision deficiency) that make it difficult or impossible to distinguish between shades of blue and green.
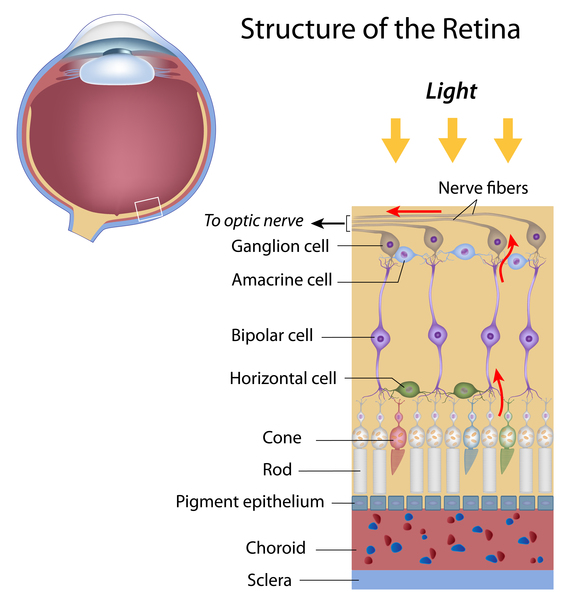
In the early stages of the condition, individuals with optic atrophy type 1 experience a progressive loss of certain cells within the retina, which is a specialized light-sensitive tissue that lines the back of the eye. The loss of these cells (known as retinal ganglion cells) is followed by the degeneration (atrophy) of the nerves that relay visual information from the eye to the brain (optic nerves), which results in further vision loss. Atrophy causes these nerves to have an abnormally pale appearance (pallor), which can be seen during an eye examination.
Frequency
Optic atrophy type 1 is estimated to affect 1 in 35,000 people worldwide. This condition is more common in Denmark, where it affects approximately 1 in 10,000 people.
Causes
Optic atrophy type 1 is caused by mutations in the OPA1 gene. The protein produced from this gene is made in cells and tissues throughout the body. The OPA1 protein is found within mitochondria, which are the energy-producing centers of cells. The protein plays a key role in the organization of the shape and structure of the mitochondria and in controlled cell death (apoptosis). The OPA1 protein is also involved in a process called oxidative phosphorylation, from which cells derive much of their energy. Additionally, the protein plays a role in the maintenance of the DNA within mitochondria, called mitochondrial DNA (mtDNA).
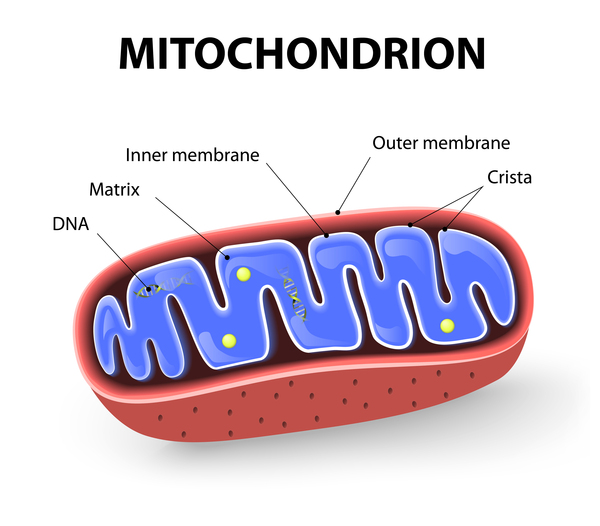
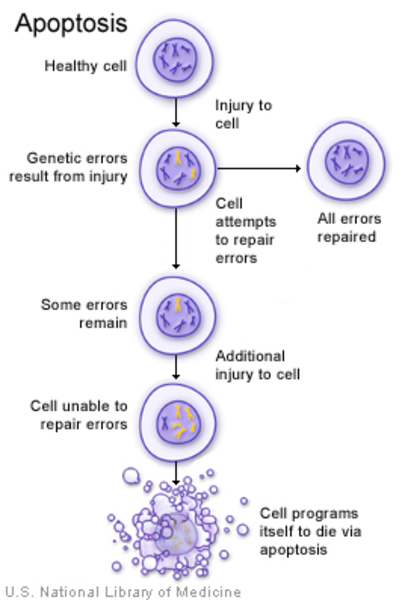

Mutations in the OPA1 gene lead to problems with mitochondrial function. The mitochondria become misshapen and disorganized and have reduced energy-producing capabilities. The maintenance of mtDNA may also be impaired, resulting in mtDNA mutations that further interfere with mitochondrial energy production. Cells that contain these poorly functioning mitochondria are more susceptible to apoptosis. In particular, cells that have high energy demands, such as retinal ganglion cells, die over time. Specialized extensions of retinal ganglion cells, called axons, form the optic nerves, so when retinal ganglion cells die, the optic nerves atrophy and cannot transmit visual information to the brain.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In most cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


In rare cases, people who have an OPA1 gene mutation do not develop optic atrophy type 1, a situation known as reduced penetrance.
Other Names for This Condition
- ADOA
- Autosomal dominant optic atrophy
- Autosomal dominant optic atrophy Kjer type
- DOA
- Dominant optic atrophy
- Kjer type optic atrophy
- Kjer's optic atrophy
- Optic atrophy, autosomal dominant
- Optic atrophy, hereditary, autosomal dominant
- Optic atrophy, juvenile
- Optic atrophy, Kjer type
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Delettre-Cribaillet C, Hamel CP, Lenaers G. Optic Atrophy Type 1 - RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. 2007 Jul 13 [updated 2015 Nov 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1248/ Citation on PubMed
- Ferre M, Bonneau D, Milea D, Chevrollier A, Verny C, Dollfus H, Ayuso C, Defoort S, Vignal C, Zanlonghi X, Charlin JF, Kaplan J, Odent S, Hamel CP, Procaccio V, Reynier P, Amati-Bonneau P. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum Mutat. 2009 Jul;30(7):E692-705. doi: 10.1002/humu.21025. Citation on PubMed
- Formichi P, Radi E, Giorgi E, Gallus GN, Brunetti J, Battisti C, Rufa A, Dotti MT, Franceschini R, Bracci L, Federico A. Analysis of opa1 isoforms expression and apoptosis regulation in autosomal dominant optic atrophy (ADOA) patients with mutations in the opa1 gene. J Neurol Sci. 2015 Apr 15;351(1-2):99-108. doi: 10.1016/j.jns.2015.02.047. Epub 2015 Mar 6. Citation on PubMed
- Roubertie A, Leboucq N, Picot MC, Nogue E, Brunel H, Le Bars E, Manes G, Angebault Prouteau C, Blanchet C, Mondain M, Chevassus H, Amati-Bonneau P, Sarzi E, Pages M, Villain M, Meunier I, Lenaers G, Hamel CP. Neuroradiological findings expand the phenotype of OPA1-related mitochondrial dysfunction. J Neurol Sci. 2015 Feb 15;349(1-2):154-60. doi: 10.1016/j.jns.2015.01.008. Epub 2015 Jan 13. Citation on PubMed
- Yu-Wai-Man P, Griffiths PG, Burke A, Sellar PW, Clarke MP, Gnanaraj L, Ah-Kine D, Hudson G, Czermin B, Taylor RW, Horvath R, Chinnery PF. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology. 2010 Aug;117(8):1538-46, 1546.e1. doi: 10.1016/j.ophtha.2009.12.038. Epub 2010 Apr 24. Citation on PubMed or Free article on PubMed Central
- Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, Wissinger B, Pinti M, Cossarizza A, Vidoni S, Valentino ML, Rugolo M, Carelli V. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008 Feb;131(Pt 2):352-67. doi: 10.1093/brain/awm335. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.