

Description
Omenn syndrome is an inherited disorder of the immune system (immunodeficiency). Omenn syndrome is one of several forms of severe combined immunodeficiency (SCID), a group of disorders that cause individuals to have virtually no immune protection from bacteria, viruses, and fungi. Individuals with SCID are prone to repeated and persistent infections that can be very serious or life-threatening. Infants with Omenn syndrome typically experience pneumonia and chronic diarrhea. Often the organisms that cause infection in people with this disorder are described as opportunistic because they ordinarily do not cause illness in healthy people.
In addition to immunodeficiency, children with Omenn syndrome develop autoimmunity, in which the immune system attacks the body's own tissues and organs. This abnormal immune reaction can cause very red skin (erythroderma), hair loss (alopecia), and an enlarged liver and spleen (hepatosplenomegaly). In addition, affected individuals have enlargement of tissues that produce infection-fighting white blood cells called lymphocytes. These include the thymus, which is a gland located behind the breastbone, and lymph nodes, which are found throughout the body.
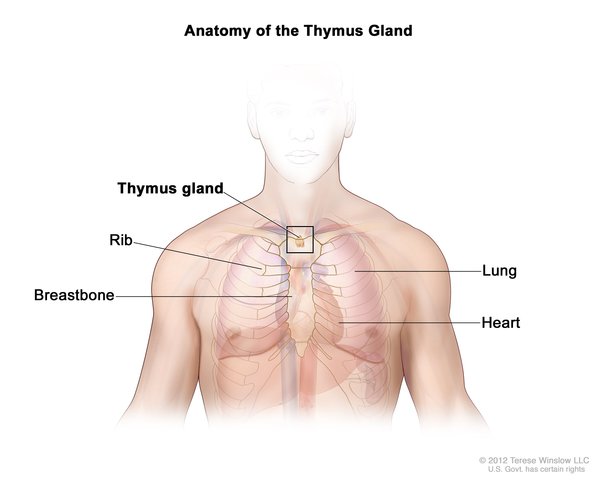
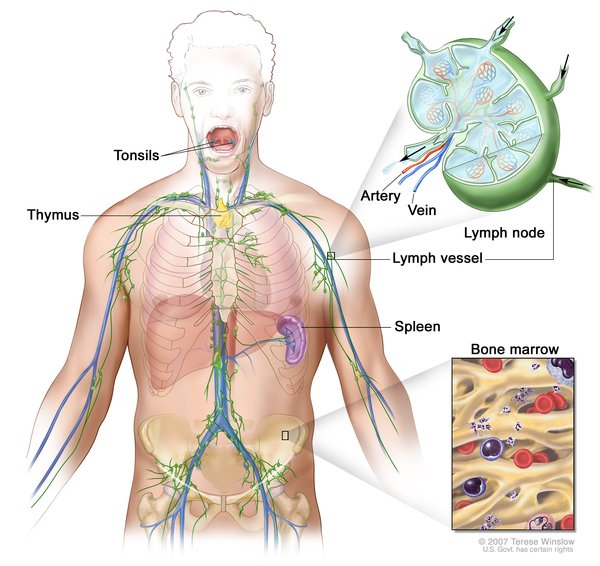
If not treated in a way that restores immune function, children with Omenn syndrome usually survive only until age 1 or 2.
Frequency
Overall, the various forms of SCID are estimated to affect 1 in 75,000 to 100,000 newborns. The exact prevalence of Omenn syndrome is unknown.
Causes
Mutations in several genes involved in immune system function can cause Omenn syndrome. The two most frequent causes are mutations in the RAG1 and RAG2 genes. These genes provide instructions for making proteins that are active in two types of lymphocytes called B cells and T cells. To help fight infections, B cells and T cells have special proteins on their surface that help them recognize foreign invaders; these proteins must be somewhat different from each other to be able to recognize a wide variety of substances. The RAG1 and RAG2 proteins help increase the diversity of proteins that are on the surface of these cells.
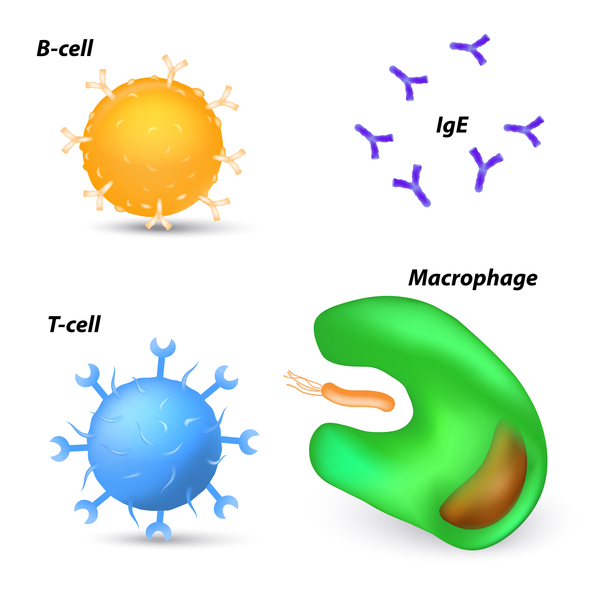
RAG1 and RAG2 gene mutations that cause Omenn syndrome drastically reduce the respective protein's function. As a result, the diversity of proteins on the surface of B cells and T cells is severely limited, impairing the cells' ability to recognize foreign invaders and fight infections. The abnormal B cells and T cells result in the frequent, life-threatening infections of Omenn syndrome. The decrease in lymphocyte function leads to a reduction in the numbers of B cells. The number of T cells is typically normal, although they are highly similar because they are derived from just a few functional precursor cells. The abnormal T cells attack the body's own cells and tissues, accounting for the autoimmune features of Omenn syndrome.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Familial reticuloendotheliosis
- Histiocytic medullary reticulosis
- Omenn's syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Franco JL, Gaspar HB, Holland SM, Klein C, Nonoyama S, Ochs HD, Oksenhendler E, Picard C, Puck JM, Sullivan K, Tang ML. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014 Apr 22;5:162. doi: 10.3389/fimmu.2014.00162. eCollection 2014. Citation on PubMed or Free article on PubMed Central
- Alsmadi O, Al-Ghonaium A, Al-Muhsen S, Arnaout R, Al-Dhekri H, Al-Saud B, Al-Kayal F, Al-Saud H, Al-Mousa H. Molecular analysis of T-B-NK+ severe combined immunodeficiency and Omenn syndrome cases in Saudi Arabia. BMC Med Genet. 2009 Nov 13;10:116. doi: 10.1186/1471-2350-10-116. Citation on PubMed or Free article on PubMed Central
- Bai X, Liu J, Zhang Z, Liu C, Zhang Y, Tang W, Dai R, Wu J, Tang X, Zhang Y, Ding Y, Jiang L, Zhao X. Clinical, immunologic, and genetic characteristics of RAG mutations in 15 Chinese patients with SCID and Omenn syndrome. Immunol Res. 2016 Apr;64(2):497-507. doi: 10.1007/s12026-015-8723-4. Citation on PubMed
- Cassani B, Poliani PL, Moratto D, Sobacchi C, Marrella V, Imperatori L, Vairo D, Plebani A, Giliani S, Vezzoni P, Facchetti F, Porta F, Notarangelo LD, Villa A, Badolato R. Defect of regulatory T cells in patients with Omenn syndrome. J Allergy Clin Immunol. 2010 Jan;125(1):209-16. doi: 10.1016/j.jaci.2009.10.023. Citation on PubMed
- Ege M, Ma Y, Manfras B, Kalwak K, Lu H, Lieber MR, Schwarz K, Pannicke U. Omenn syndrome due to ARTEMIS mutations. Blood. 2005 Jun 1;105(11):4179-86. doi: 10.1182/blood-2004-12-4861. Epub 2005 Feb 24. Citation on PubMed
- Sharapova SO, Guryanova IE, Pashchenko OE, Kondratenko IV, Kostyuchenko LV, Rodina YA, Varlamova TV, Bondarenko AV, Chernyshova LI, Gyseva MN, Belevtsev MV, Minakovskaya NV, Aleinikova OV. Molecular Characteristics, Clinical and Immunologic Manifestations of 11 Children with Omenn Syndrome in East Slavs (Russia, Belarus, Ukraine). J Clin Immunol. 2016 Jan;36(1):46-55. doi: 10.1007/s10875-015-0216-7. Epub 2015 Nov 23. Citation on PubMed
- Somech R, Simon AJ, Lev A, Dalal I, Spirer Z, Goldstein I, Nagar M, Amariglio N, Rechavi G, Roifman CM. Reduced central tolerance in Omenn syndrome leads to immature self-reactive oligoclonal T cells. J Allergy Clin Immunol. 2009 Oct;124(4):793-800. doi: 10.1016/j.jaci.2009.06.048. Epub 2009 Sep 19. Citation on PubMed
- Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008 Dec;122(6):1082-6. doi: 10.1016/j.jaci.2008.09.037. Epub 2008 Nov 6. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.