

Description
Myosin storage myopathy is a condition that affects muscle (myopathy). This disorder causes muscle weakness that can worsen slowly over time. The signs and symptoms of myosin storage myopathy often begin in childhood, although they can begin later. The specific features of myosin storage myopathy and the severity of the condition can vary, even among members of the same family.
Children with myosin storage myopathy may start walking later than usual and have a waddling gait. They can have trouble climbing stairs and difficulty lifting their arms above shoulder level. Weak shoulder muscles often make the shoulder blades (scapulae) "stick out" from the back, a sign known as scapular winging. Affected individuals may also have a spine that curves to the side (scoliosis). Some individuals can also develop weakness of the muscles that control breathing (respiratory insufficiency) or weakness of the heart (cardiac) muscle.

Myosin storage myopathy belongs to a group of disorders called congenital myopathies. The signs and symptoms of congenital myopathies are often present at birth (congenital) or soon after. Congenital myopathies may affect the muscles used for movement (skeletal muscle) and, less commonly, the cardiac muscle.
Frequency
Myosin storage myopathy is rare, although the exact number of people with this condition is unknown.
Causes
Genetic changes that cause disease are called pathogenic variants. Pathogenic variants in the MYH7 gene cause myosin storage myopathy. The MYH7 gene provides instructions for making a protein known as myosin-7. This protein is found in cardiac muscle and in certain skeletal muscle fibers.
Myosin-7 is the major component of the thick filament in muscle cell structures called sarcomeres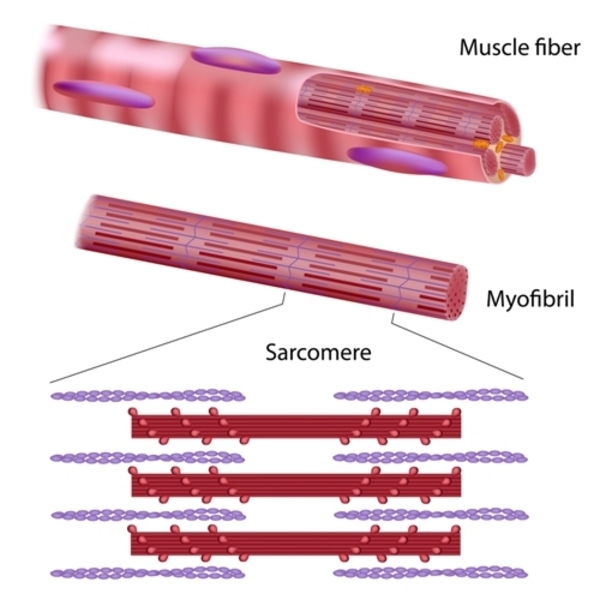 . Sarcomeres are the basic units of muscle contraction. They are composed of overlapping thick and thin filaments that undergo cycles of attachment and release. These cycles allow the thin filaments to slide past the thick filaments, which shortens the sarcomere and causes the muscle to contract.
. Sarcomeres are the basic units of muscle contraction. They are composed of overlapping thick and thin filaments that undergo cycles of attachment and release. These cycles allow the thin filaments to slide past the thick filaments, which shortens the sarcomere and causes the muscle to contract.
Pathogenic variants in the MYH7 gene can cause cells to produce a version of myosin-7 that does not function properly. The altered proteins accumulate within the skeletal muscle fibers, forming protein clumps that can be seen when muscle tissue is removed for examination (muscle biopsy). It is unclear exactly how these changes lead to the muscle weakness seen in people with myosin storage myopathy.
Inheritance
Myosin storage myopathy is typically inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In rare cases, myosin storage myopathy has followed an autosomal recessive pattern , which means both copies of the gene in each cell must have a pathogenic variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a pathogenic variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Hyaline body myopathy
- MYH7-related skeletal myopathy
- Myosin storage congenital myopathy
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Armel TZ, Leinwand LA. Mutations in the beta-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc Natl Acad Sci U S A. 2009 Apr 14;106(15):6291-6. doi: 10.1073/pnas.0900107106. Epub 2009 Mar 31. Citation on PubMed or Free article on PubMed Central
- Bahout M, Severa G, Kamoun E, Bouhour F, Pegat A, Toutain A, Lagrange E, Duval F, Tard C, De la Cruz E, Feasson L, Jacquin-Piques A, Richard P, Metay C, Cavalli M, Romero NB, Evangelista T, Sole G, Carlier RY, Laforet P, Acket B, Behin A, Fernandez-Eulate G, Leonard-Louis S, Quijano-Roy S, Pereon Y, Salort-Campana E, Nadaj-Pakleza A, Masingue M, Malfatti E, Stojkovic T, Villar-Quiles RN. MYH7-related myopathies: clinical, myopathological and genotypic spectrum in a multicentre French cohort. J Neurol Neurosurg Psychiatry. 2025 Apr 10;96(5):453-461. doi: 10.1136/jnnp-2024-334263. Citation on PubMed
- Beecroft SJ, van de Locht M, de Winter JM, Ottenheijm CA, Sewry CA, Mohammed S, Ryan MM, Woodcock IR, Sanders L, Gooding R, Davis MR, Oates EC, Laing NG, Ravenscroft G, McLean CA, Jungbluth H. Recessive MYH7-related myopathy in two families. Neuromuscul Disord. 2019 Jun;29(6):456-467. doi: 10.1016/j.nmd.2019.04.002. Epub 2019 Apr 12. Citation on PubMed
- Cancilla PA, Kalyanaraman K, Verity MA, Munsat T, Pearson CM. Familial myopathy with probable lysis of myofibrils in type I fibers. Neurology. 1971 Jun;21(6):579-85. doi: 10.1212/wnl.21.6.579. No abstract available. Citation on PubMed
- Claeys KG. Congenital myopathies: an update. Dev Med Child Neurol. 2020 Mar;62(3):297-302. doi: 10.1111/dmcn.14365. Epub 2019 Oct 2. Citation on PubMed
- Dahl-Halvarsson M, Pokrzywa M, Rauthan M, Pilon M, Tajsharghi H. Myosin Storage Myopathy in C. elegans and Human Cultured Muscle Cells. PLoS One. 2017 Jan 26;12(1):e0170613. doi: 10.1371/journal.pone.0170613. eCollection 2017. Citation on PubMed
- Fiorillo C, Astrea G, Savarese M, Cassandrini D, Brisca G, Trucco F, Pedemonte M, Trovato R, Ruggiero L, Vercelli L, D'Amico A, Tasca G, Pane M, Fanin M, Bello L, Broda P, Musumeci O, Rodolico C, Messina S, Vita GL, Sframeli M, Gibertini S, Morandi L, Mora M, Maggi L, Petrucci A, Massa R, Grandis M, Toscano A, Pegoraro E, Mercuri E, Bertini E, Mongini T, Santoro L, Nigro V, Minetti C, Santorelli FM, Bruno C; Italian Network on Congenital Myopathies. MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis. 2016 Jul 7;11(1):91. doi: 10.1186/s13023-016-0476-1. Citation on PubMed
- Gao Y, Peng L, Zhao C. MYH7 in cardiomyopathy and skeletal muscle myopathy. Mol Cell Biochem. 2024 Feb;479(2):393-417. doi: 10.1007/s11010-023-04735-x. Epub 2023 Apr 20. Citation on PubMed
- Li N, Zhao Z, Shen H, Bing Q, Guo X, Hu J. MYH7 mutation associated with two phenotypes of myopathy. Neurol Sci. 2018 Feb;39(2):333-339. doi: 10.1007/s10072-017-3192-2. Epub 2017 Nov 24. Citation on PubMed
- Naderi N, Mohsen-Pour N, Nilipour Y, Pourirahim M, Maleki M, Kalayinia S. A novel heterozygous missense MYH7 mutation potentially causes an autosomal dominant form of myosin storage myopathy with dilated cardiomyopathy. BMC Cardiovasc Disord. 2023 Oct 4;23(1):487. doi: 10.1186/s12872-023-03538-8. Citation on PubMed
- Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013 Jan;125(1):3-18. doi: 10.1007/s00401-012-1024-2. Epub 2012 Aug 5. Citation on PubMed or Free article on PubMed Central
- Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol. 2003 Oct;54(4):494-500. doi: 10.1002/ana.10693. Citation on PubMed
- Viswanathan MC, Tham RC, Kronert WA, Sarsoza F, Trujillo AS, Cammarato A, Bernstein SI. Myosin storage myopathy mutations yield defective myosin filament assembly in vitro and disrupted myofibrillar structure and function in vivo. Hum Mol Genet. 2017 Dec 15;26(24):4799-4813. doi: 10.1093/hmg/ddx359. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.