

Description
Multiple sulfatase deficiency is a condition that mainly affects the brain, skin, and skeleton. Because the signs and symptoms of multiple sulfatase deficiency vary widely, researchers have split the condition into three types: neonatal, late-infantile, and juvenile.
The neonatal type is the most severe form, with signs and symptoms appearing soon after birth. Affected individuals have deterioration of tissue in the nervous system (leukodystrophy), which can contribute to movement problems, seizures, developmental delay, and slow growth. They also have dry, scaly skin (ichthyosis) and excess hair growth (hypertrichosis). Skeletal abnormalities can include abnormal side-to-side curvature of the spine (scoliosis), joint stiffness, and dysostosis multiplex, which refers to a specific pattern of skeletal abnormalities seen on x-ray. Individuals with the neonatal type typically have facial features that can be described as "coarse." Affected individuals may also have hearing loss, heart malformations, and an enlarged liver and spleen (hepatosplenomegaly). Many of the signs and symptoms of neonatal multiple sulfatase deficiency worsen over time.

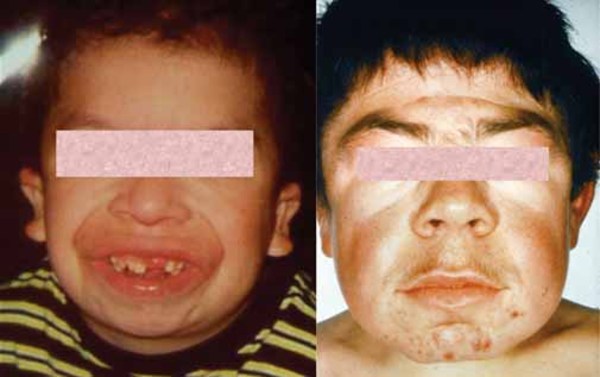

The late-infantile type is the most common form of multiple sulfatase deficiency. It is characterized by normal cognitive development in early childhood followed by a progressive loss of mental abilities and movement (psychomotor regression) due to leukodystrophy or other brain abnormalities. Individuals with this form of the condition do not have as many features as those with the neonatal type, but they often have ichthyosis, skeletal abnormalities, and coarse facial features.
The juvenile type is the rarest form of multiple sulfatase deficiency. Signs and symptoms of the juvenile type appear in mid- to late childhood. Affected individuals have normal early cognitive development but then experience psychomotor regression; however, the regression in the juvenile type usually occurs at a slower rate than in the late-infantile type. Ichthyosis is also common in the juvenile type of multiple sulfatase deficiency.
Life expectancy is shortened in individuals with all types of multiple sulfatase deficiency. Typically, affected individuals survive only a few years after the signs and symptoms of the condition appear, but life expectancy varies depending on the severity of the condition and how quickly the neurological problems worsen.
Frequency
Multiple sulfatase deficiency is estimated to occur in 1 per million individuals worldwide. More than 140 cases have been reported in the scientific literature.
Causes
Multiple sulfatase deficiency is caused by mutations in the SUMF1 gene. This gene provides instructions for making an enzyme called formylglycine-generating enzyme (FGE). This enzyme is found in a cell structure called the endoplasmic reticulum, which is involved in protein processing and transport. The FGE enzyme modifies other enzymes called sulfatases, which aid in breaking down substances that contain chemical groups known as sulfates. These substances include a variety of sugars, fats, and hormones.

Most SUMF1 gene mutations severely reduce the function of the FGE enzyme or lead to the production of an unstable enzyme that is quickly broken down. The activity of multiple sulfatases is impaired because the FGE enzyme modifies all known sulfatase enzymes. Sulfate-containing molecules that are not broken down build up in cells, often resulting in cell death. The death of cells in particular tissues, specifically the brain, skeleton, and skin, cause many of the signs and symptoms of multiple sulfatase deficiency.
Research indicates that mutations that lead to reduced FGE enzyme function are associated with the less severe cases of the condition, whereas mutations that lead to the production of an unstable FGE enzyme tend to be associated with the more severe cases of multiple sulfatase deficiency.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Austin syndrome
- Juvenile sulfatidosis, Austin type
- MSD
- Mucosulfatidosis
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ahrens-Nicklas R, Schlotawa L, Ballabio A, Brunetti-Pierri N, De Castro M, Dierks T, Eichler F, Ficicioglu C, Finglas A, Gaertner J, Kirmse B, Klepper J, Lee M, Olsen A, Parenti G, Vossough A, Vanderver A, Adang LA. Complex care of individuals with multiple sulfatase deficiency: Clinical cases and consensus statement. Mol Genet Metab. 2018 Mar;123(3):337-346. doi: 10.1016/j.ymgme.2018.01.005. Epub 2018 Jan 31. Citation on PubMed
- Annunziata I, Bouche V, Lombardi A, Settembre C, Ballabio A. Multiple sulfatase deficiency is due to hypomorphic mutations of the SUMF1 gene. Hum Mutat. 2007 Sep;28(9):928. doi: 10.1002/humu.9504. Citation on PubMed
- Blanco-Aguirre ME, Kofman-Alfaro SH, Rivera-Vega MR, Medina C, Valdes-Flores M, Rizzo WB, Cuevas-Covarrubias SA. Unusual clinical presentation in two cases of multiple sulfatase deficiency. Pediatr Dermatol. 2001 Sep-Oct;18(5):388-92. doi: 10.1046/j.1525-1470.2001.01959.x. Citation on PubMed
- Cosma MP, Pepe S, Parenti G, Settembre C, Annunziata I, Wade-Martins R, Di Domenico C, Di Natale P, Mankad A, Cox B, Uziel G, Mancini GM, Zammarchi E, Donati MA, Kleijer WJ, Filocamo M, Carrozzo R, Carella M, Ballabio A. Molecular and functional analysis of SUMF1 mutations in multiple sulfatase deficiency. Hum Mutat. 2004 Jun;23(6):576-81. doi: 10.1002/humu.20040. Citation on PubMed
- Diaz-Font A, Santamaria R, Cozar M, Blanco M, Chamoles N, Coll MJ, Chabas A, Vilageliu L, Grinberg D. Clinical and mutational characterization of three patients with multiple sulfatase deficiency: report of a new splicing mutation. Mol Genet Metab. 2005 Sep-Oct;86(1-2):206-11. doi: 10.1016/j.ymgme.2005.07.004. Citation on PubMed
- Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, von Figura K. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell. 2003 May 16;113(4):435-44. doi: 10.1016/s0092-8674(03)00347-7. Citation on PubMed
- Incecik F, Ozbek MN, Gungor S, Pepe S, Herguner OM, Mungan NO, Gungor S, Altunbasak S. Multiple sulfatase deficiency: A case series of four children. Ann Indian Acad Neurol. 2013 Oct;16(4):720-2. doi: 10.4103/0972-2327.120449. Citation on PubMed or Free article on PubMed Central
- Schlotawa L, Ennemann EC, Radhakrishnan K, Schmidt B, Chakrapani A, Christen HJ, Moser H, Steinmann B, Dierks T, Gartner J. SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. Eur J Hum Genet. 2011 Mar;19(3):253-61. doi: 10.1038/ejhg.2010.219. Epub 2011 Jan 12. Citation on PubMed or Free article on PubMed Central
- Schlotawa L, Preiskorn J, Ahrens-Nicklas R, Schiller S, Adang LA, Gartner J, Friede T. A systematic review and meta-analysis of published cases reveals the natural disease history in multiple sulfatase deficiency. J Inherit Metab Dis. 2020 Nov;43(6):1288-1297. doi: 10.1002/jimd.12282. Epub 2020 Jul 22. Citation on PubMed
- Schlotawa L, Radhakrishnan K, Baumgartner M, Schmid R, Schmidt B, Dierks T, Gartner J. Rapid degradation of an active formylglycine generating enzyme variant leads to a late infantile severe form of multiple sulfatase deficiency. Eur J Hum Genet. 2013 Sep;21(9):1020-3. doi: 10.1038/ejhg.2012.291. Epub 2013 Jan 16. Citation on PubMed or Free article on PubMed Central
- Schlotawa L, Steinfeld R, von Figura K, Dierks T, Gartner J. Molecular analysis of SUMF1 mutations: stability and residual activity of mutant formylglycine-generating enzyme determine disease severity in multiple sulfatase deficiency. Hum Mutat. 2008 Jan;29(1):205. doi: 10.1002/humu.9515. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.