

Description
Multiple mitochondrial dysfunctions syndrome is characterized by impairment of cellular structures called mitochondria, which are the energy-producing centers of cells. While certain mitochondrial disorders are caused by impairment of a single stage of energy production, individuals with multiple mitochondrial dysfunctions syndrome have reduced function of more than one stage. The signs and symptoms of this severe condition begin early in life, and affected individuals usually do not live past infancy.
Affected infants typically have severe brain dysfunction (encephalopathy), which can contribute to weak muscle tone (hypotonia), seizures, and delayed development of mental and movement abilities (psychomotor delay). These infants often have difficulty growing and gaining weight at the expected rate (failure to thrive). Most affected babies have a buildup of a chemical called lactic acid in the body (lactic acidosis), which can be life-threatening. They may also have high levels of a molecule called glycine (hyperglycinemia) or elevated levels of sugar (hyperglycemia) in the blood. Some babies with multiple mitochondrial dysfunctions syndrome have high blood pressure in the blood vessels that connect to the lungs (pulmonary hypertension) or weakening of the heart muscle (cardiomyopathy).
Frequency
Multiple mitochondrial dysfunctions syndrome is a rare condition; its prevalence is unknown. It is one of several conditions classified as mitochondrial disorders, which affect an estimated 1 in 5,000 people worldwide.
Causes
Multiple mitochondrial dysfunctions syndrome can be caused by mutations in the NFU1 or BOLA3 gene. The proteins produced from each of these genes appear to be involved in the formation of molecules called iron-sulfur (Fe-S) clusters or in the attachment of these clusters to other proteins. Certain proteins require attachment of Fe-S clusters to function properly.
The NFU-1 and BOLA3 proteins play an important role in mitochondria. In these structures, several proteins carry out a series of chemical steps to convert the energy in food into a form that cells can use. Many of the proteins involved in these steps require Fe-S clusters to function, including protein complexes called complex I, complex II, and complex III.
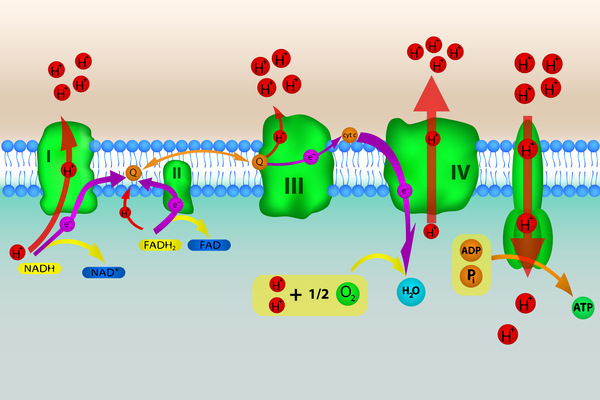
Fe-S clusters are also required for another mitochondrial protein to function; this protein is involved in the modification of additional proteins that aid in energy production in mitochondria, including the pyruvate dehydrogenase complex and the alpha-ketoglutarate dehydrogenase complex (also known as the oxoglutarate dehydrogenase complex). This modification is also critical to the function of the glycine cleavage system, a set of proteins that breaks down a protein building block (amino acid) called glycine when levels become too high.
Mutations in the NFU1 or BOLA3 gene reduce or eliminate production of the respective protein, which impairs Fe-S cluster formation. Consequently, proteins affected by the presence of Fe-S clusters, including those involved in energy production and glycine breakdown, cannot function normally. Reduced activity of complex I, II, or III, pyruvate dehydrogenase, or alpha-ketoglutarate dehydrogenase leads to potentially fatal lactic acidosis, encephalopathy, and other signs and symptoms of multiple mitochondrial dysfunctions syndrome. In some affected individuals, impairment of the glycine cleavage system leads to a buildup of glycine.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- MMDS
- Multiple mitochondrial dysfunction syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong WH, Ogilvie I, Shoubridge EA, Robinson BH. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011 Oct 7;89(4):486-95. doi: 10.1016/j.ajhg.2011.08.011. Epub 2011 Sep 22. Citation on PubMed or Free article on PubMed Central
- Haack TB, Rolinski B, Haberberger B, Zimmermann F, Schum J, Strecker V, Graf E, Athing U, Hoppen T, Wittig I, Sperl W, Freisinger P, Mayr JA, Strom TM, Meitinger T, Prokisch H. Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings. J Inherit Metab Dis. 2013 Jan;36(1):55-62. doi: 10.1007/s10545-012-9489-7. Epub 2012 May 5. Citation on PubMed
- Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, Del Toro M, Labayru MT, Landa J, Font A, Garcia-Villoria J, Merinero B, Ugarte M, Gutierrez-Solana LG, Campistol J, Garcia-Cazorla A, Vaquerizo J, Riudor E, Briones P, Elpeleg O, Ribes A, Lill R. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011 Nov 11;89(5):656-67. doi: 10.1016/j.ajhg.2011.10.005. Citation on PubMed or Free article on PubMed Central
- Seyda A, Newbold RF, Hudson TJ, Verner A, MacKay N, Winter S, Feigenbaum A, Malaney S, Gonzalez-Halphen D, Cuthbert AP, Robinson BH. A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am J Hum Genet. 2001 Feb;68(2):386-96. doi: 10.1086/318196. Epub 2001 Jan 10. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.