

Description
Mucopolysaccharidosis type II (MPS II), also known as Hunter syndrome, is a condition that affects many different parts of the body. The condition occurs almost exclusively in boys, although it has been reported in a few girls. It is a progressively debilitating disorder; however, the rate of progression varies among affected individuals.
At birth, individuals with MPS II do not display any features of the condition. Between ages 2 and 4, they develop full lips; large, rounded cheeks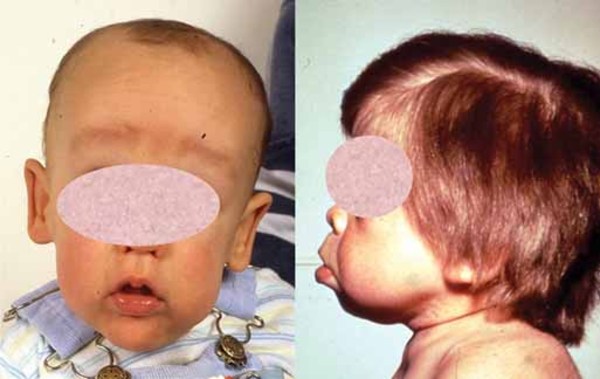 ; a broad nose
; a broad nose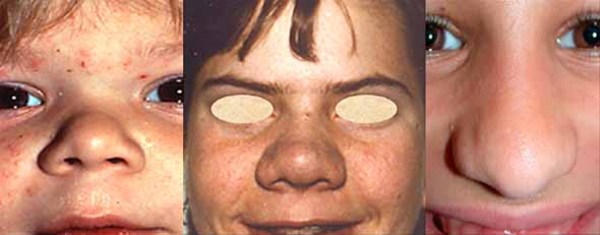 ; and an enlarged tongue (macroglossia
; and an enlarged tongue (macroglossia ). The vocal cords also enlarge, which results in a deep, hoarse voice. Narrowing of the airway causes frequent upper respiratory infections and short pauses in breathing during sleep (sleep apnea). As the disorder progresses, individuals need medical assistance to keep their airway open.
). The vocal cords also enlarge, which results in a deep, hoarse voice. Narrowing of the airway causes frequent upper respiratory infections and short pauses in breathing during sleep (sleep apnea). As the disorder progresses, individuals need medical assistance to keep their airway open.
Many other organs and tissues are affected in people with MPS II. Individuals with this disorder often have a large head (macrocephaly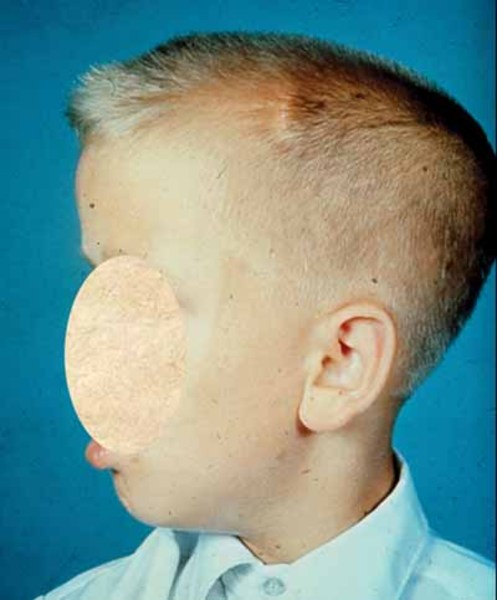 ), a buildup of fluid in the brain (hydrocephalus), a short neck, an enlarged liver and spleen (hepatosplenomegaly), and a soft out-pouching around the belly-button (umbilical hernia
), a buildup of fluid in the brain (hydrocephalus), a short neck, an enlarged liver and spleen (hepatosplenomegaly), and a soft out-pouching around the belly-button (umbilical hernia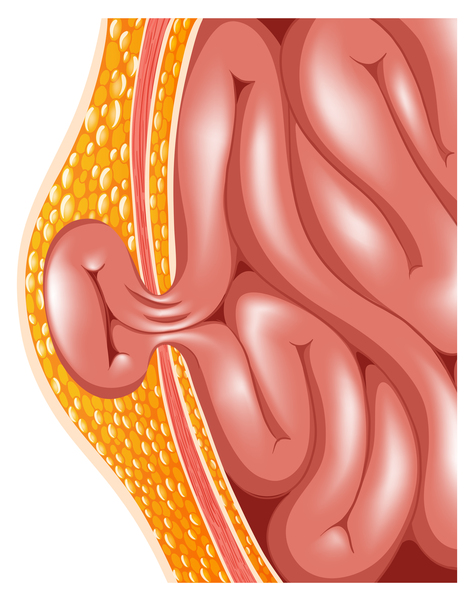 ) or lower abdomen (inguinal hernia). People with MPS II usually have thick skin that is not very stretchy. Some affected individuals also have distinctive white skin growths that look like pebbles. Most people with this disorder develop hearing loss. Some individuals with MPS II develop problems with the light-sensitive tissue in the back of the eye (retina
) or lower abdomen (inguinal hernia). People with MPS II usually have thick skin that is not very stretchy. Some affected individuals also have distinctive white skin growths that look like pebbles. Most people with this disorder develop hearing loss. Some individuals with MPS II develop problems with the light-sensitive tissue in the back of the eye (retina ) and have reduced vision. Carpal tunnel syndrome commonly occurs in children with this disorder and is characterized by numbness, tingling, and weakness in the hand and fingers. Narrowing of the spinal canal (spinal stenosis
) and have reduced vision. Carpal tunnel syndrome commonly occurs in children with this disorder and is characterized by numbness, tingling, and weakness in the hand and fingers. Narrowing of the spinal canal (spinal stenosis ) in the neck can compress and damage the spinal cord. The heart is also significantly affected by MPS II, and many individuals develop heart valve
) in the neck can compress and damage the spinal cord. The heart is also significantly affected by MPS II, and many individuals develop heart valve problems. Heart valve abnormalities can cause the heart to become enlarged (ventricular hypertrophy) and can eventually lead to abnormalities in the heart's rhythm (arrhythmia) and heart failure.
problems. Heart valve abnormalities can cause the heart to become enlarged (ventricular hypertrophy) and can eventually lead to abnormalities in the heart's rhythm (arrhythmia) and heart failure.
Children with MPS II grow steadily until about age 5, and then their growth slows and they develop short stature. Individuals with this condition have joint deformities (contractures) that significantly affect mobility. Most people with MPS II also have dysostosis multiplex, which refers to multiple skeletal abnormalities that can be seen on x-rays. Dysostosis multiplex includes a generalized thickening of certain bones, particularly the ribs.
There are two types of MPS II: the neuropathic form, which is more severe, and the non-neuropathic form, which is less severe. While both types affect many different organs and tissues as described above, people with neuropathic MPS II also experience a decline in intellectual function and a more rapid disease progression. Individuals with this form begin to lose basic functional skills (developmentally regress) between the ages of 6 and 8. Their life expectancy is 10 to 20 years. Individuals with non-neuropathic MPS II also have a shortened lifespan, but they typically live into adulthood, and their intelligence is not affected. Heart disease and airway obstruction are major causes of death in people with both types of MPS II.
Frequency
MPS II occurs in approximately 1 in 100,000 to 1 in 170,000 males.
Causes
Variants (also called mutations) in the IDS gene cause MPS II. The IDS gene provides instructions for producing the I2S enzyme, which is involved in the breakdown of large sugar molecules called glycosaminoglycans (GAGs). GAGs were originally called mucopolysaccharides, which is where this condition gets its name.
IDS gene variants that cause MPS II reduce or completely eliminate the function of the I2S enzyme. Lack of I2S enzyme activity leads to the accumulation of GAGs within cells, specifically inside the lysosomes. Lysosomes are compartments in the cell that digest and recycle different types of molecules. Conditions that cause molecules to build up inside the lysosomes, including MPS II, are called lysosomal storage disorders.
are compartments in the cell that digest and recycle different types of molecules. Conditions that cause molecules to build up inside the lysosomes, including MPS II, are called lysosomal storage disorders.
The accumulation of GAGs increases the size of the lysosomes, which is why many tissues and organs are enlarged in people with this disorder. Researchers believe that the accumulated GAGs may also interfere with the functions of other proteins inside the lysosomes and disrupt the movement of molecules inside the cell. In addition, buildup in lysosomes may trigger the release of molecules called cytokines that stimulate inflammation and may contribute to the progression of MPS II.
Inheritance
This condition is inherited in an X-linked recessive pattern . The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes
. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes . In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a variant would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that both copies of a gene would be altered, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a variant would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that both copies of a gene would be altered, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Other Names for This Condition
- Hunter syndrome
- I2S deficiency
- Iduronate 2-sulfatase deficiency
- MPS II
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bhalla A, Ravi R, Fang M, Arguello A, Davis SS, Chiu CL, Blumenfeld JR, Nguyen HN, Earr TK, Wang J, Astarita G, Zhu Y, Fiore D, Scearce-Levie K, Diaz D, Cahan H, Troyer MD, Harris JM, Escolar ML. Characterization of Fluid Biomarkers Reveals Lysosome Dysfunction and Neurodegeneration in Neuronopathic MPS II Patients. Int J Mol Sci. 2020 Jul 22;21(15):5188. doi: 10.3390/ijms21155188. Citation on PubMed
- Clarke LA. The mucopolysaccharidoses: a success of molecular medicine. Expert Rev Mol Med. 2008 Jan 18;10:e1. doi: 10.1017/S1462399408000550. Citation on PubMed
- Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Munoz V, Muenzer J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008 Feb;121(2):e377-86. doi: 10.1542/peds.2007-1350. Citation on PubMed
- Ream MA, Lam WKK, Grosse SD, Ojodu J, Jones E, Prosser LA, Rose AM, Comeau AM, Tanksley S, Powell CM, Kemper AR. Evidence and recommendation for mucopolysaccharidosis type II newborn screening in the United States. Genet Med. 2023 Feb;25(2):100330. doi: 10.1016/j.gim.2022.10.012. Epub 2022 Nov 29. Citation on PubMed
- Scarpa M, Lampe C. Mucopolysaccharidosis Type II. 2007 Nov 6 [updated 2026 Apr 28]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1274/ Citation on PubMed
- Trowbridge JM, Gallo RL. Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology. 2002 Sep;12(9):117R-25R. doi: 10.1093/glycob/cwf066. Citation on PubMed
- Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol. 2005 Apr;32(4):270-2. doi: 10.1016/j.pediatrneurol.2004.10.009. Citation on PubMed
- Tylki-Szymanska A. Mucopolysaccharidosis type II, Hunter's syndrome. Pediatr Endocrinol Rev. 2014 Sep;12 Suppl 1:107-13. Citation on PubMed
- Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, Meldgaard Lund A, Malm G, Van der Ploeg AT, Zeman J. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008 Mar;167(3):267-77. doi: 10.1007/s00431-007-0635-4. Epub 2007 Nov 23. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.