

Description
MN1 C-terminal truncation (MCTT) syndrome is a condition characterized by intellectual disability, developmental delay, distinctive facial features, and brain abnormalities.
Most people with MCTT syndrome have mild to moderate intellectual disability. Many affected individuals are nonverbal, but some have speech limited to one or two words or communicate using sign language. Most children with this condition have delayed development of motor skills, such as crawling or walking, but are able to walk by age 2 or 3. However, they often need help with fine-motor skills, such as getting dressed or using a fork when eating.
Individuals with MCTT syndrome often have distinctive facial features that include a sunken appearance of the middle of the face (midface hypoplasia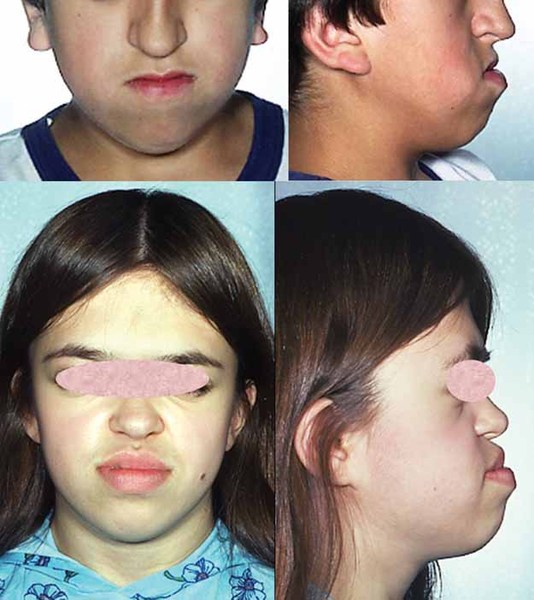 ); a high arch in the roof of the mouth (high-arched palate
); a high arch in the roof of the mouth (high-arched palate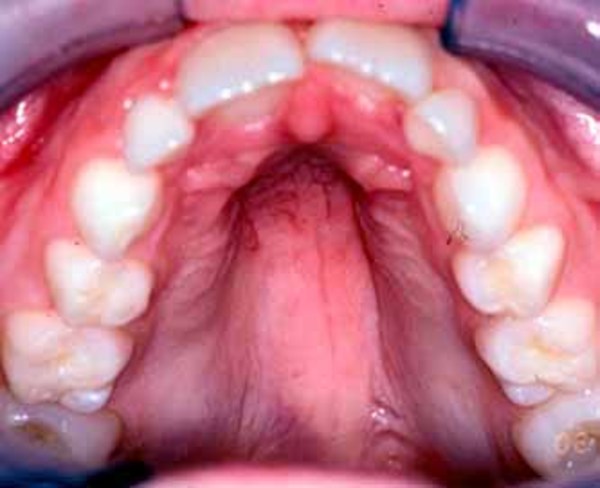 ); outside corners of the eyes that point downward (downslanting palpebral fissures
); outside corners of the eyes that point downward (downslanting palpebral fissures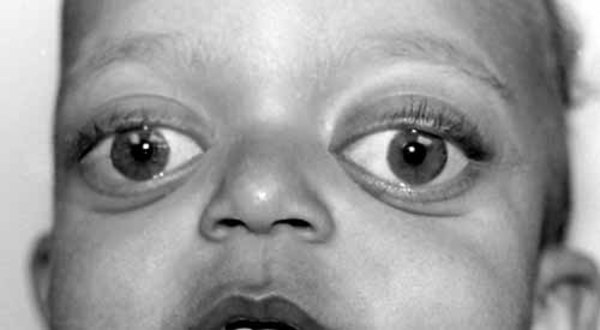 ); widely spaced eyes (hypertelorism
); widely spaced eyes (hypertelorism ); shallow and bulging eyes (exophthalmos); a short, upturned nose
); shallow and bulging eyes (exophthalmos); a short, upturned nose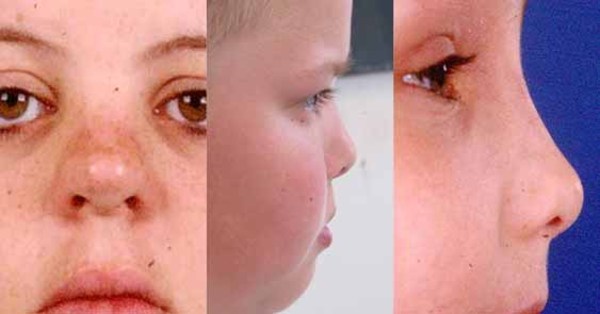 ; and
small, low-set ears
; and
small, low-set ears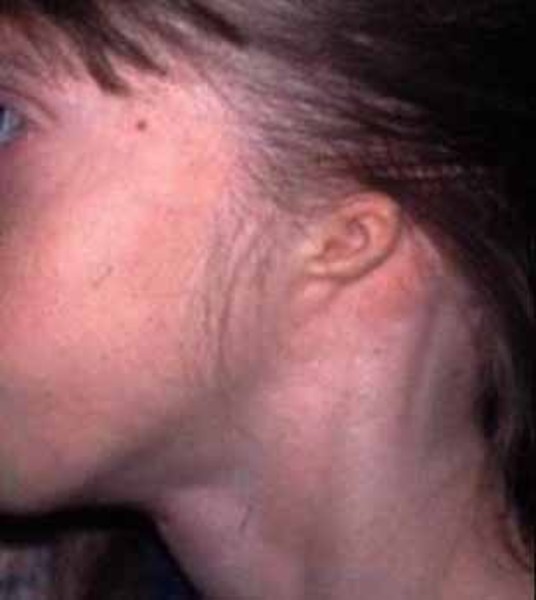 . Some affected individuals have dental abnormalities, such as cone-shaped (conical
. Some affected individuals have dental abnormalities, such as cone-shaped (conical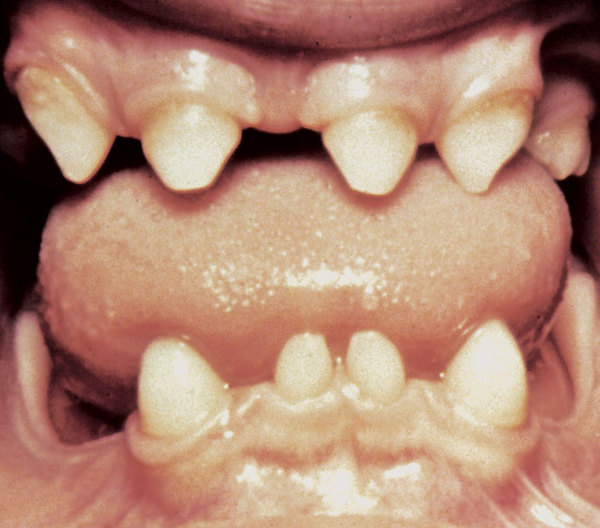 ), jagged, or crowded teeth. Rarely, people with MCTT syndrome have premature fusion of certain skull bones (craniosynostosis).
), jagged, or crowded teeth. Rarely, people with MCTT syndrome have premature fusion of certain skull bones (craniosynostosis).
People with MCTT syndrome often have characteristic brain abnormalities. The
surface of the brain normally has many ridges or folds, called gyri. A common brain abnormality in people with MCTT syndrome is called perisylvian polymicrogyria, in which an area of the brain called the perisylvian region develops too many gyri, and the folds are irregular and unusually small. Individuals with MCTT syndrome can also have a malformation of the part of the brain that coordinates movement (the cerebellum
normally has many ridges or folds, called gyri. A common brain abnormality in people with MCTT syndrome is called perisylvian polymicrogyria, in which an area of the brain called the perisylvian region develops too many gyri, and the folds are irregular and unusually small. Individuals with MCTT syndrome can also have a malformation of the part of the brain that coordinates movement (the cerebellum ). This malformation, called
atypical rhombencephalosynapsis
). This malformation, called
atypical rhombencephalosynapsis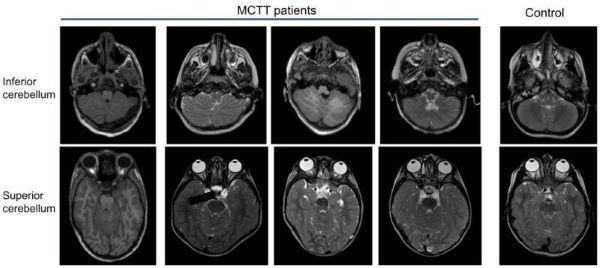 ,
is characterized by tissue loss in the central part of the cerebellum (known as the vermis) and fusion of the two sides of the cerebellum. These brain abnormalities likely contribute to the movement problems and intellectual disability that are common in MCTT syndrome.
,
is characterized by tissue loss in the central part of the cerebellum (known as the vermis) and fusion of the two sides of the cerebellum. These brain abnormalities likely contribute to the movement problems and intellectual disability that are common in MCTT syndrome.
Less common features of MCTT syndrome include hearing loss, seizures, abnormal curvature of the spine, and heart abnormalities.
Frequency
The prevalence of MCTT syndrome is unknown, although it is thought to be a rare disorder. At least 25 affected individuals have been described in the scientific literature.
Causes
MCTT syndrome is caused by mutations in the MN1 gene. This gene provides instructions for making a protein whose function is unclear. Based on its interaction with other proteins, the MN1 protein is thought to play a role in regulating the activity of other genes, particularly those needed for the development of the skull and brain.
All MN1 gene mutations that cause MCTT syndrome occur near the end (terminal) portion of the gene. As a result, an abnormally short (truncated) protein is produced. These mutations are reflected in the condition name, MN1 C-terminal truncation syndrome.
Research shows that a shortened MN1 protein cannot interact with other proteins, leading to a buildup of the abnormal MN1 protein in the cell nucleus. It is likely that without the normal function of the MN1 protein, the activity of certain genes involved in the development of the skull and brain is unregulated, leading to the signs and symptoms of MCTT syndrome.
Inheritance
MCTT syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Most cases of this condition result from new (de novo) mutations in the MN1 gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- CEBALID
- Craniofacial defects, dysmorphic ears, structural brain abnormalities, expressive language delay, and impaired intellectual development
- MCTT syndrome
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Mak CCY, Doherty D, Lin AE, Vegas N, Cho MT, Viot G, Dimartino C, Weisfeld-Adams JD, Lessel D, Joss S, Li C, Gonzaga-Jauregui C, Zarate YA, Ehmke N, Horn D, Troyer C, Kant SG, Lee Y, Ishak GE, Leung G, Barone Pritchard A, Yang S, Bend EG, Filippini F, Roadhouse C, Lebrun N, Mehaffey MG, Martin PM, Apple B, Millan F, Puk O, Hoffer MJV, Henderson LB, McGowan R, Wentzensen IM, Pei S, Zahir FR, Yu M, Gibson WT, Seman A, Steeves M, Murrell JR, Luettgen S, Francisco E, Strom TM, Amlie-Wolf L, Kaindl AM, Wilson WG, Halbach S, Basel-Salmon L, Lev-El N, Denecke J, Vissers LELM, Radtke K, Chelly J, Zackai E, Friedman JM, Bamshad MJ, Nickerson DA; University of Washington Center for Mendelian Genomics; Reid RR, Devriendt K, Chae JH, Stolerman E, McDougall C, Powis Z, Bienvenu T, Tan TY, Orenstein N, Dobyns WB, Shieh JT, Choi M, Waggoner D, Gripp KW, Parker MJ, Stoler J, Lyonnet S, Cormier-Daire V, Viskochil D, Hoffman TL, Amiel J, Chung BHY, Gordon CT. MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis. Brain. 2020 Jan 1;143(1):55-68. doi: 10.1093/brain/awz379. Erratum In: Brain. 2020 Mar 1;143(3):e24. doi: 10.1093/brain/awaa007. Citation on PubMed
- Mak CCY, Fung JLF, Lee M, Lin AE, Amiel J, Doherty D, Gordon CT, Chung BHY. MN1 C-Terminal Truncation Syndrome. 2020 Aug 13. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK560443/ Citation on PubMed
- Miyake N, Takahashi H, Nakamura K, Isidor B, Hiraki Y, Koshimizu E, Shiina M, Sasaki K, Suzuki H, Abe R, Kimura Y, Akiyama T, Tomizawa SI, Hirose T, Hamanaka K, Miyatake S, Mitsuhashi S, Mizuguchi T, Takata A, Obo K, Kato M, Ogata K, Matsumoto N. Gain-of-Function MN1 Truncation Variants Cause a Recognizable Syndrome with Craniofacial and Brain Abnormalities. Am J Hum Genet. 2020 Jan 2;106(1):13-25. doi: 10.1016/j.ajhg.2019.11.011. Epub 2019 Dec 12. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.