

Description
Miyoshi myopathy is a muscle disorder that begins with weakness in the muscles that are located away from the center of the body (distal muscles), such as those in the legs. During early to mid-adulthood, affected individuals typically begin to experience muscle weakness and wasting (atrophy) in one or both calves. If only one leg is affected, the calves appear different in size (asymmetrical). Calf weakness can make it difficult to stand on tiptoe.
As Miyoshi myopathy slowly worsens, the muscle weakness and atrophy spread up the leg to the muscles in the thigh and buttock and can also involve the upper arm and shoulder muscles. Eventually, affected individuals may have difficulty climbing stairs or walking for an extended period of time. Some people with Miyoshi myopathy may eventually need wheelchair assistance.
Rarely, abnormal heart rhythms (arrhythmias) have developed in people with Miyoshi myopathy. Individuals with Miyoshi myopathy have highly elevated levels of an enzyme called creatine kinase (CK) in their blood, which often indicates muscle disease.
Frequency
The exact prevalence of Miyoshi myopathy is unknown. In Japan, where the condition was first described, it is estimated to affect 1 in 440,000 individuals.
Causes
Miyoshi myopathy is caused by mutations in the DYSF or ANO5 gene. When Miyoshi myopathy is caused by ANO5 gene mutations it is sometimes referred to as distal anoctaminopathy; when this condition is caused by DYSF gene mutations it is known as a dysferlinopathy. The DYSF and ANO5 genes provide instructions for making proteins primarily found in muscles that are used for movement (skeletal muscles). The protein produced from the DYSF gene, called dysferlin, is found in the thin membrane called the sarcolemma that surrounds muscle fibers. Dysferlin is thought to aid in repairing the sarcolemma when it becomes damaged or torn due to muscle strain.
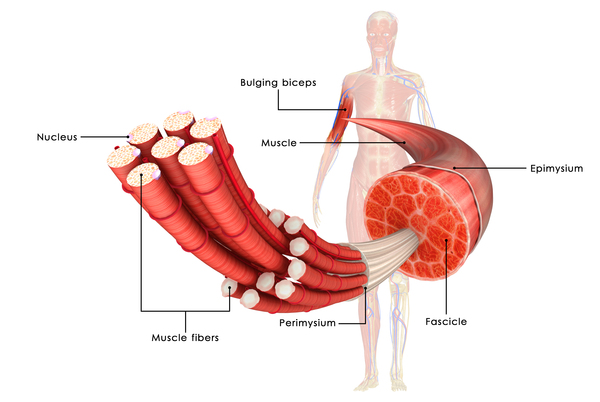
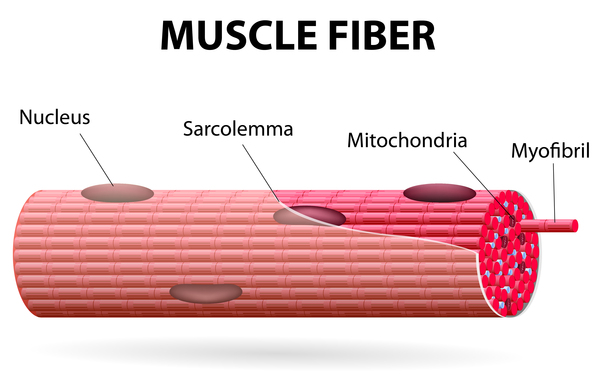
The ANO5 gene provides instructions for making a protein called anoctamin-5. This protein is located within the membrane of a cell structure called the endoplasmic reticulum, which is involved in protein production, processing, and transport. Anoctamin-5 is thought to act as a channel, allowing charged chlorine atoms (chloride ions) to flow in and out of the endoplasmic reticulum. The regulation of chloride flow within muscle cells plays a role in controlling muscle tensing (contraction) and relaxation.


DYSF and ANO5 gene mutations often result in a decrease or elimination of the corresponding protein. A lack of dysferlin leads to a reduced ability to repair damage done to the sarcolemma of muscle fibers. As a result, damage accumulates and leads to atrophy of the muscle fiber. It is unclear why this damage leads to the specific pattern of weakness and atrophy that is characteristic of Miyoshi myopathy. The effects of the loss of anoctamin-5 are also unclear. While chloride is necessary for normal muscle function, it is unknown how a lack of this chloride channel causes the signs and symptoms of Miyoshi myopathy.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Distal muscular dystrophy, Miyoshi type
- Miyoshi distal myopathy
- Miyoshi muscular dystrophy
- MMD
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Amato AA, Brown RH Jr. Dysferlinopathies. Handb Clin Neurol. 2011;101:111-8. doi: 10.1016/B978-0-08-045031-5.00007-4. Citation on PubMed
- Bouquet F, Cossee M, Behin A, Deburgrave N, Romero N, Leturcq F, Eymard B. Miyoshi-like distal myopathy with mutations in anoctamin 5 gene. Rev Neurol (Paris). 2012 Feb;168(2):135-41. doi: 10.1016/j.neurol.2011.10.005. Epub 2012 Feb 13. Citation on PubMed
- Fanin M, Angelini C. Progress and challenges in diagnosis of dysferlinopathy. Muscle Nerve. 2016 Nov;54(5):821-835. doi: 10.1002/mus.25367. Citation on PubMed
- Liewluck T, Winder TL, Dimberg EL, Crum BA, Heppelmann CJ, Wang Y, Bergen HR 3rd, Milone M. ANO5-muscular dystrophy: clinical, pathological and molecular findings. Eur J Neurol. 2013 Oct;20(10):1383-9. doi: 10.1111/ene.12191. Epub 2013 May 12. Citation on PubMed
- Linssen WH, de Voogt WG, Krahn M, Bernard R, Levy N, Wokke JH, Ginjaar HB, de Visser M. Long-term follow-up study on patients with Miyoshi phenotype of distal muscular dystrophy. Eur J Neurol. 2013 Jun;20(6):968-74. doi: 10.1111/ene.12129. Epub 2013 Mar 26. Citation on PubMed
- Nguyen K, Bassez G, Bernard R, Krahn M, Labelle V, Figarella-Branger D, Pouget J, Hammouda el H, Beroud C, Urtizberea A, Eymard B, Leturcq F, Levy N. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum Mutat. 2005 Aug;26(2):165. doi: 10.1002/humu.9355. Citation on PubMed
- Nilsson MI, Laureano ML, Saeed M, Tarnopolsky MA. Dysferlin aggregation in limb-girdle muscular dystrophy type 2B/Miyoshi Myopathy necessitates mutational screen for diagnosis [corrected]. Muscle Nerve. 2013 May;47(5):740-7. doi: 10.1002/mus.23666. Epub 2013 Mar 21. Citation on PubMed
- Penttila S, Palmio J, Suominen T, Raheem O, Evila A, Muelas Gomez N, Tasca G, Waddell LB, Clarke NF, Barboi A, Hackman P, Udd B. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology. 2012 Mar 20;78(12):897-903. doi: 10.1212/WNL.0b013e31824c4682. Epub 2012 Mar 7. Citation on PubMed
- Ten Dam L, van der Kooi AJ, Rovekamp F, Linssen WH, de Visser M. Comparing clinical data and muscle imaging of DYSF and ANO5 related muscular dystrophies. Neuromuscul Disord. 2014 Dec;24(12):1097-102. doi: 10.1016/j.nmd.2014.07.004. Epub 2014 Aug 1. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.