

Description
Mitochondrial neurogastrointestinal encephalopathy (MNGIE) disease is a condition that affects several parts of the body, particularly the digestive system and nervous system. The major features of MNGIE disease can appear anytime from infancy to adulthood, but signs and symptoms most often begin by age 20. The medical problems associated with this disorder worsen over time.
Almost all people with MNGIE disease have a condition known as gastrointestinal dysmotility, in which the muscles and nerves of the digestive system do not move food through the digestive tract efficiently. The resulting digestive problems include feelings of fullness (satiety) after eating only a small amount, trouble swallowing (dysphagia), nausea and vomiting after eating, episodes of abdominal pain, diarrhea, and intestinal blockage. These gastrointestinal problems lead to extreme weight loss and reduced muscle mass (cachexia).
MNGIE disease is also characterized by abnormalities of the nervous system, although these tend to be milder than the gastrointestinal problems. Affected individuals experience tingling, numbness, and weakness in their limbs (peripheral neuropathy), particularly in the hands and feet. Additional neurological signs and symptoms can include droopy eyelids (ptosis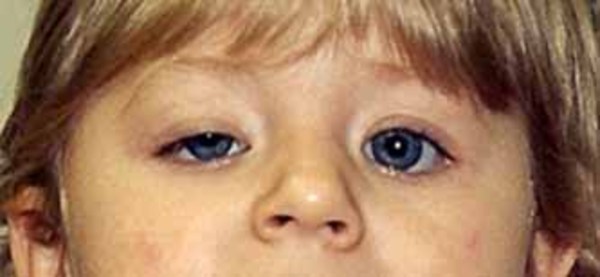 ), weakness of the muscles that control eye movement (ophthalmoplegia), and hearing loss. Leukoencephalopathy, which is the deterioration of a type of brain tissue known as white matter, is a hallmark of MNGIE disease. These changes in the brain can be seen with magnetic resonance imaging (MRI), though they usually do not cause symptoms in people with this disorder.
), weakness of the muscles that control eye movement (ophthalmoplegia), and hearing loss. Leukoencephalopathy, which is the deterioration of a type of brain tissue known as white matter, is a hallmark of MNGIE disease. These changes in the brain can be seen with magnetic resonance imaging (MRI), though they usually do not cause symptoms in people with this disorder.
Frequency
The prevalence of MNGIE disease is unknown. Fewer than 200 people with this disorder have been described in the scientific literature.
Causes
Variants (also called mutations) in the TYMP gene cause most cases of MNGIE disease. The TYMP gene provides instructions for making an enzyme called thymidine phosphorylase. Thymidine is a molecule known as a nucleoside. After a chemical modification, thymidine is used as a building block of DNA . Thymidine phosphorylase breaks down thymidine into smaller molecules, which helps regulate the level of nucleosides in cells.
. Thymidine phosphorylase breaks down thymidine into smaller molecules, which helps regulate the level of nucleosides in cells.
TYMP gene variants greatly reduce or eliminate the activity of thymidine phosphorylase. A shortage of this enzyme allows thymidine to build up to very high levels in the body. Researchers believe that this excess of thymidine damages a particular kind of DNA known as mitochondrial DNA or mtDNA. Mitochondria are structures within cells that convert the energy from food into a form that cells can use. Although most DNA is packaged in chromosomes within the nucleus, mitochondria also have a small amount of their own DNA.
Mitochondria use nucleosides, including thymidine, to build new molecules of mtDNA. A loss of thymidine phosphorylase activity and the resulting buildup of thymidine disrupt the usual maintenance and repair of mtDNA. As a result, variants can accumulate in mtDNA, causing it to become unstable. In people with MNGIE disease, mitochondria may also have less mtDNA than usual (mtDNA depletion). These genetic changes impair the normal function of mitochondria. Although mtDNA abnormalities underlie the digestive and neurological problems characteristic of MNGIE disease, it is unclear how defective mitochondria cause the specific features of the disorder.
Rarely, variants in other genes can cause MNGIE disease. In the remaining cases, the cause of the condition is unknown.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- MEPOP
- Mitochondrial myopathy with sensorimotor polyneuropathy, ophthalmoplegia, and pseudo-obstruction
- Mitochondrial neurogastrointestinal encephalopathy syndrome
- MNGIE disease
- MNGIE syndrome
- Myoneurogastrointestinal encephalopathy syndrome
- Oculogastrointestinal muscular dystrophy
- OGIMD
- POLIP
- Polyneuropathy, ophthalmoplegia, leukoencephalopathy, and intestinal pseudo-obstruction
- Thymidine phosphorylase deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Hirano M, Marti R, Spinazzola A, Nishino I, Nishigaki Y. Thymidine phosphorylase deficiency causes MNGIE: an autosomal recessive mitochondrial disorder. Nucleosides Nucleotides Nucleic Acids. 2004 Oct;23(8-9):1217-25. doi: 10.1081/NCN-200027485. Citation on PubMed
- Hirano M, Silvestri G, Blake DM, Lombes A, Minetti C, Bonilla E, Hays AP, Lovelace RE, Butler I, Bertorini TE, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. 1994 Apr;44(4):721-7. doi: 10.1212/wnl.44.4.721. Citation on PubMed
- Hirano M. Mitochondrial Neurogastrointestinal Encephalopathy Disease. 2005 Apr 22 [updated 2016 Jan 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1179/ Citation on PubMed
- Lara MC, Valentino ML, Torres-Torronteras J, Hirano M, Marti R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): biochemical features and therapeutic approaches. Biosci Rep. 2007 Jun;27(1-3):151-63. doi: 10.1007/s10540-007-9043-2. Citation on PubMed
- Marti R, Spinazzola A, Nishino I, Andreu AL, Naini A, Tadesse S, Oliver JA, Hirano M. Mitochondrial neurogastrointestinal encephalomyopathy and thymidine metabolism: results and hypotheses. Mitochondrion. 2002 Nov;2(1-2):143-7. doi: 10.1016/s1567-7249(02)00036-3. Citation on PubMed
- Nishino I, Spinazzola A, Hirano M. MNGIE: from nuclear DNA to mitochondrial DNA. Neuromuscul Disord. 2001 Jan;11(1):7-10. doi: 10.1016/s0960-8966(00)00159-0. Citation on PubMed
- Nishino I, Spinazzola A, Papadimitriou A, Hammans S, Steiner I, Hahn CD, Connolly AM, Verloes A, Guimaraes J, Maillard I, Hamano H, Donati MA, Semrad CE, Russell JA, Andreu AL, Hadjigeorgiou GM, Vu TH, Tadesse S, Nygaard TG, Nonaka I, Hirano I, Bonilla E, Rowland LP, DiMauro S, Hirano M. Mitochondrial neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol. 2000 Jun;47(6):792-800. Citation on PubMed
- Rajakulendran S, Pitceathly RD, Taanman JW, Costello H, Sweeney MG, Woodward CE, Jaunmuktane Z, Holton JL, Jacques TS, Harding BN, Fratter C, Hanna MG, Rahman S. A Clinical, Neuropathological and Genetic Study of Homozygous A467T POLG-Related Mitochondrial Disease. PLoS One. 2016 Jan 6;11(1):e0145500. doi: 10.1371/journal.pone.0145500. eCollection 2016. Citation on PubMed
- Teitelbaum JE, Berde CB, Nurko S, Buonomo C, Perez-Atayde AR, Fox VL. Diagnosis and management of MNGIE syndrome in children: case report and review of the literature. J Pediatr Gastroenterol Nutr. 2002 Sep;35(3):377-83. doi: 10.1097/00005176-200209000-00029. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.