

Description
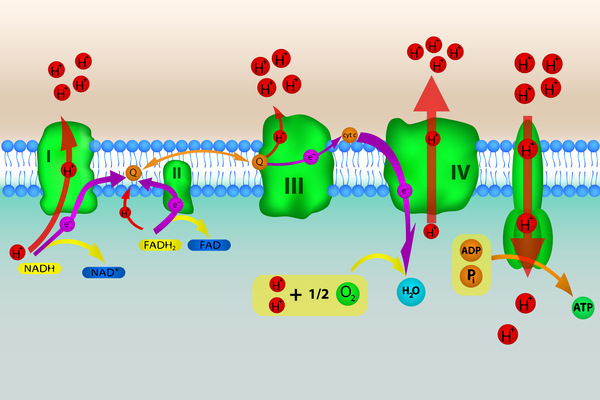
Mitochondrial complex V deficiency is a shortage (deficiency) of a protein complex called complex V or a loss of its function. Complex V is found in cell structures called mitochondria, which convert the energy from food into a form that cells can use. Complex V is the last of five mitochondrial complexes that carry out a multistep process called oxidative phosphorylation, through which cells derive much of their energy.

Mitochondrial complex V deficiency can cause a wide variety of signs and symptoms affecting many organs and systems of the body, particularly the nervous system and the heart. The disorder can be life-threatening in infancy or early childhood. Affected individuals may have feeding problems, slow growth, low muscle tone (hypotonia), extreme fatigue (lethargy), and developmental delay. They tend to develop elevated levels of lactic acid in the blood (lactic acidosis), which can cause nausea, vomiting, weakness, and rapid breathing. High levels of ammonia in the blood (hyperammonemia) can also occur in affected individuals, and in some cases result in abnormal brain function (encephalopathy) and damage to other organs.
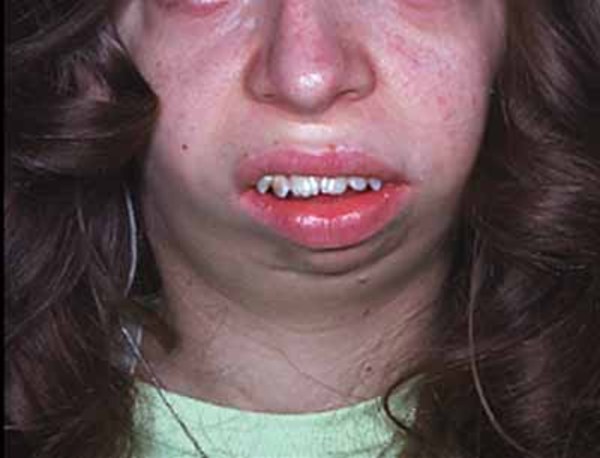
Another common feature of mitochondrial complex V deficiency is hypertrophic cardiomyopathy. This condition is characterized by thickening (hypertrophy) of the heart (cardiac) muscle that can lead to heart failure. People with mitochondrial complex V deficiency may also have a characteristic pattern of facial features, including a high forehead, curved eyebrows, outside corners of the eyes that point downward (downslanting palpebral fissures), a prominent bridge of the nose, low-set ears, thin lips, and a small chin (micrognathia).
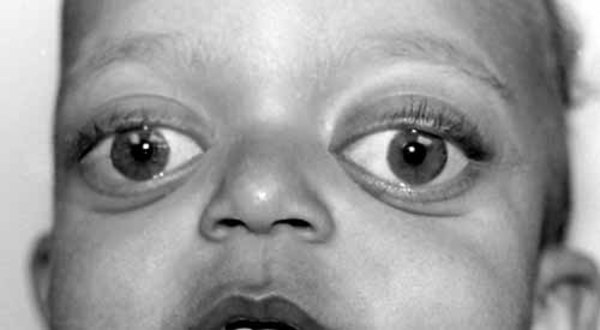
Some people with mitochondrial complex V deficiency have groups of signs and symptoms that are classified as a specific syndrome. For example, mitochondrial complex V deficiency can cause a condition called neuropathy, ataxia, and retinitis pigmentosa (NARP). NARP causes a variety of signs and symptoms chiefly affecting the nervous system. Beginning in childhood or early adulthood, most people with NARP experience numbness, tingling, or pain in the arms and legs (sensory neuropathy); muscle weakness; and problems with balance and coordination (ataxia). Many affected individuals also have cognitive impairment and an eye disorder called retinitis pigmentosa that causes vision loss.
A condition called Leigh syndrome can also be caused by mitochondrial complex V deficiency. Leigh syndrome is characterized by progressive loss of mental and movement abilities (developmental or psychomotor regression) and typically results in death within 2 to 3 years after the onset of symptoms. Both NARP and Leigh syndrome can also have other causes.
Frequency
The prevalence of mitochondrial complex V deficiency is unknown. Researchers suggest that the condition may be underdiagnosed because affected individuals can have a wide variety of features that are not specific to this condition.
Causes
Mutations in any of several genes can cause mitochondrial complex V deficiency. These genes provide instructions for making components of complex V or proteins that help assemble the complex.
Gene mutations that cause mitochondrial complex V deficiency impair the formation or function of complex V. As a result, complex V activity is reduced and oxidative phosphorylation is impaired. Researchers believe that problems with oxidative phosphorylation can lead to cell death by reducing the amount of energy available in the cell. It is thought that tissues and organs that require a lot of energy, such as the nervous system, the heart, the liver, the kidneys, and the muscles used for movement (skeletal muscles), are most affected by a reduction in oxidative phosphorylation.
Some genes known to be involved in mitochondrial complex V deficiency are found in nuclear DNA, which is packaged in chromosomes within the cell nucleus. TMEM70 is the nuclear gene most commonly mutated in mitochondrial complex V deficiency. This gene provides instructions for making a protein called transmembrane protein 70, which is thought to play an important role in assembling and stabilizing complex V. Mutations in the TMEM70 gene reduce the amount of complex V that is formed, leading to the signs and symptoms of mitochondrial complex V deficiency.
Other genes involved in mitochondrial complex V deficiency are found in mitochondrial DNA (mtDNA), which is located in the mitochondria themselves. Most of the body's cells contain many mitochondria, and the mitochondria each contain many sets of mtDNA. When a mutation occurs in mtDNA, either all the mtDNA will have the mutation (homoplasmy), or just some of the mtDNA will contain the change (heteroplasmy). A higher percentage of mutated mtDNA typically leads to more severe disease.
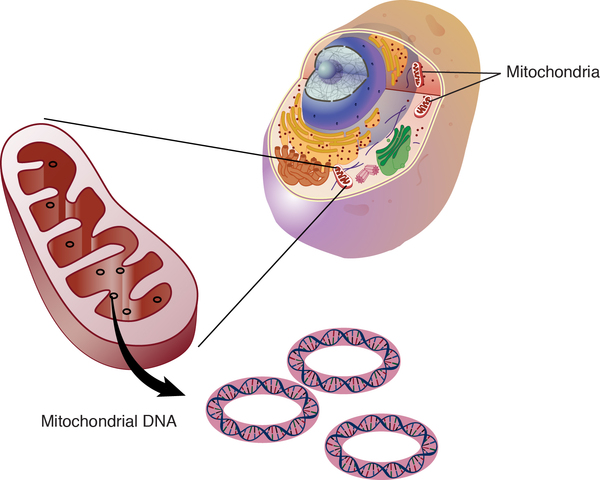
Inheritance
When mitochondrial complex V deficiency is caused by a mutation in a gene found in nuclear DNA, it has autosomal recessive inheritance. Autosomal recessive means that both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition because the other copy of the gene is normal.

When mitochondrial complex V deficiency is caused by a mutation in a gene found in mtDNA, it is inherited in a mitochondrial pattern, which is also known as maternal inheritance. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA mutations only from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
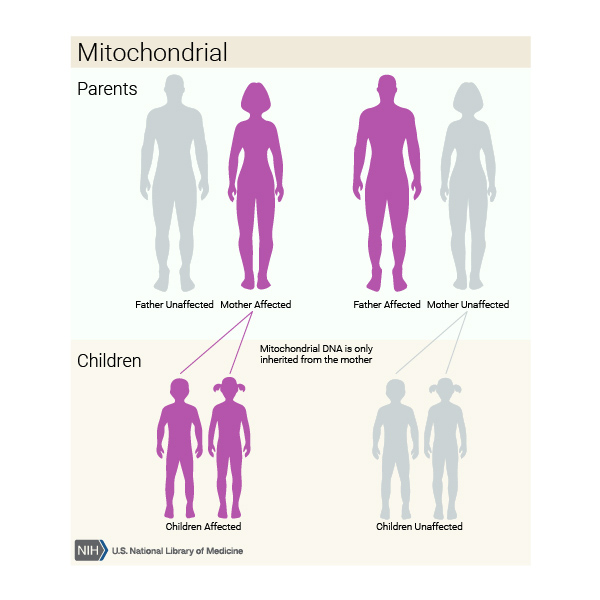
Other Names for This Condition
- ATP synthase deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017 Jan;241(2):236-250. doi: 10.1002/path.4809. Epub 2016 Nov 2. Citation on PubMed or Free article on PubMed Central
- Diodato D, Invernizzi F, Lamantea E, Fagiolari G, Parini R, Menni F, Parenti G, Bollani L, Pasquini E, Donati MA, Cassandrini D, Santorelli FM, Haack TB, Prokisch H, Ghezzi D, Lamperti C, Zeviani M. Common and Novel TMEM70 Mutations in a Cohort of Italian Patients with Mitochondrial Encephalocardiomyopathy. JIMD Rep. 2015;15:71-8. doi: 10.1007/8904_2014_300. Epub 2014 Apr 17. Citation on PubMed or Free article on PubMed Central
- Fernandez-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta. 2009 Jan;1793(1):200-11. doi: 10.1016/j.bbamcr.2008.05.028. Epub 2008 Jun 21. Citation on PubMed
- Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, Nijtmans L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017 Jan 10;25(1):128-139. doi: 10.1016/j.cmet.2016.09.002. Epub 2016 Oct 6. Citation on PubMed
- Hejzlarova K, Mracek T, Vrbacky M, Kaplanova V, Karbanova V, Nuskova H, Pecina P, Houstek J. Nuclear genetic defects of mitochondrial ATP synthase. Physiol Res. 2014;63(Suppl 1):S57-71. doi: 10.33549/physiolres.932643. Citation on PubMed
- Houstek J, Pickova A, Vojtiskova A, Mracek T, Pecina P, Jesina P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim Biophys Acta. 2006 Sep-Oct;1757(9-10):1400-5. doi: 10.1016/j.bbabio.2006.04.006. Epub 2006 Apr 19. Citation on PubMed
- Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis. 2012 Mar;35(2):211-25. doi: 10.1007/s10545-011-9382-9. Epub 2011 Aug 27. Citation on PubMed or Free article on PubMed Central
- Magner M, Dvorakova V, Tesarova M, Mazurova S, Hansikova H, Zahorec M, Brennerova K, Bzduch V, Spiegel R, Horovitz Y, Mandel H, Eminoglu FT, Mayr JA, Koch J, Martinelli D, Bertini E, Konstantopoulou V, Smet J, Rahman S, Broomfield A, Stojanovic V, Dionisi-Vici C, van Coster R, Morava E, Sperl W, Zeman J, Honzik T. TMEM70 deficiency: long-term outcome of 48 patients. J Inherit Metab Dis. 2015 May;38(3):417-26. doi: 10.1007/s10545-014-9774-8. Epub 2014 Oct 18. Citation on PubMed
- Sperl W, Jesina P, Zeman J, Mayr JA, Demeirleir L, VanCoster R, Pickova A, Hansikova H, Houst'kova H, Krejcik Z, Koch J, Smet J, Muss W, Holme E, Houstek J. Deficiency of mitochondrial ATP synthase of nuclear genetic origin. Neuromuscul Disord. 2006 Dec;16(12):821-9. doi: 10.1016/j.nmd.2006.08.008. Epub 2006 Oct 17. Citation on PubMed
- Spiegel R, Khayat M, Shalev SA, Horovitz Y, Mandel H, Hershkovitz E, Barghuti F, Shaag A, Saada A, Korman SH, Elpeleg O, Yatsiv I. TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: further delineation of a new syndrome. J Med Genet. 2011 Mar;48(3):177-82. doi: 10.1136/jmg.2010.084608. Epub 2010 Dec 8. Citation on PubMed
- Torraco A, Verrigni D, Rizza T, Meschini MC, Vazquez-Memije ME, Martinelli D, Bianchi M, Piemonte F, Dionisi-Vici C, Santorelli FM, Bertini E, Carrozzo R. TMEM70: a mutational hot spot in nuclear ATP synthase deficiency with a pivotal role in complex V biogenesis. Neurogenetics. 2012 Nov;13(4):375-86. doi: 10.1007/s10048-012-0343-8. Epub 2012 Sep 18. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.