

Description
Leigh syndrome is a severe neurological disorder that usually becomes apparent in the first year of life. This condition is characterized by progressive loss of mental and movement abilities (psychomotor regression) and typically results in death within two to three years, usually due to respiratory failure. A small number of individuals do not develop symptoms until adulthood or have symptoms that worsen more slowly.
The first signs of Leigh syndrome seen in infancy are usually vomiting, diarrhea, and difficulty swallowing (dysphagia), which disrupts eating. These problems often result in an inability to grow and gain weight at the expected rate (failure to thrive). Severe muscle and movement problems are common in Leigh syndrome. Affected individuals may develop weak muscle tone (hypotonia), involuntary muscle contractions (dystonia), and problems with movement and balance (ataxia). Loss of sensation and weakness in the limbs (peripheral neuropathy), common in people with Leigh syndrome, may also make movement difficult.
Several other features may occur in people with Leigh syndrome. Many individuals with this condition develop weakness or paralysis of the muscles that move the eyes (ophthalmoparesis); rapid, involuntary eye movements (nystagmus); or degeneration of the nerves that carry information from the eyes to the brain (optic atrophy). Severe breathing problems are common, and these problems can worsen until they cause acute respiratory failure. Some affected individuals develop hypertrophic cardiomyopathy, which is a thickening of the heart muscle that forces the heart to work harder to pump blood. In addition, a substance called lactate can build up in the body, and excessive amounts are often found in the blood, urine, or the fluid that surrounds and protects the brain and spinal cord (cerebrospinal fluid) of people with Leigh syndrome.
The signs and symptoms of Leigh syndrome are caused in part by patches of damaged tissue (lesions) that develop in the brains of people with this condition. A medical procedure called magnetic resonance imaging (MRI) reveals characteristic lesions in certain regions of the brain. These regions include the basal ganglia, which help control movement; the cerebellum, which controls the ability to balance and coordinates movement; and the brainstem, which connects the brain to the spinal cord and controls functions such as swallowing and breathing. The brain lesions are often accompanied by loss of the myelin coating around nerves (demyelination), which reduces the ability of the nerves to activate muscles used for movement or relay sensory information from the rest of the body back to the brain.


Frequency
Leigh syndrome affects at least 1 in 40,000 newborns. The condition is more common in certain populations. For example, the condition occurs in approximately 1 in 2,000 newborns in the Saguenay Lac-Saint-Jean region of Quebec, Canada and in approximately 1 in 1,700 individuals on the Faroe Islands.
Causes
Leigh syndrome can be caused by variants (also called mutations) in one of more than 110 different genes. In humans, most genes are found in DNA in the cell's nucleus , called nuclear DNA. However, some genes are found in DNA in specialized structures in the cell called mitochondria
, called nuclear DNA. However, some genes are found in DNA in specialized structures in the cell called mitochondria . This type of DNA is known as mitochondrial DNA (mtDNA). While most people with Leigh syndrome have a variant in nuclear DNA, about 20 percent have a variant in mtDNA.
. This type of DNA is known as mitochondrial DNA (mtDNA). While most people with Leigh syndrome have a variant in nuclear DNA, about 20 percent have a variant in mtDNA.
Most genes associated with Leigh syndrome are involved in the process of energy production in mitochondria. Mitochondria use oxygen to convert the energy from food into a form cells can use through a process called oxidative phosphorylation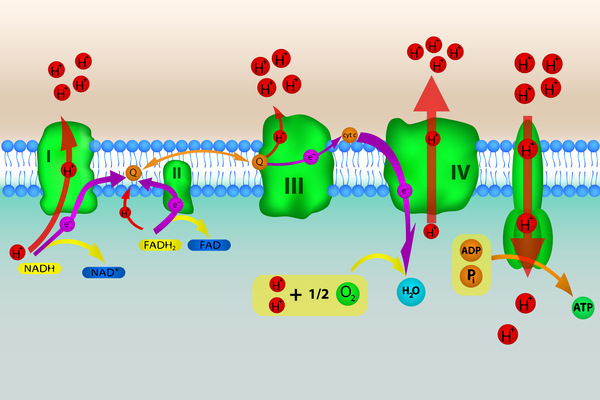 . Five protein complexes, made up of several proteins each, are involved in this process. The complexes are named complex I, complex II, complex III, complex IV, and complex V. During oxidative phosphorylation, the protein complexes drive the production of adenosine triphosphate (ATP), the cell's main energy source, through a step-by-step transfer of negatively charged particles called electrons. Many of the gene variants associated with Leigh syndrome affect proteins in these complexes or disrupt their assembly. These variants reduce or eliminate the activity of one or more of these complexes, which can lead to Leigh syndrome.
. Five protein complexes, made up of several proteins each, are involved in this process. The complexes are named complex I, complex II, complex III, complex IV, and complex V. During oxidative phosphorylation, the protein complexes drive the production of adenosine triphosphate (ATP), the cell's main energy source, through a step-by-step transfer of negatively charged particles called electrons. Many of the gene variants associated with Leigh syndrome affect proteins in these complexes or disrupt their assembly. These variants reduce or eliminate the activity of one or more of these complexes, which can lead to Leigh syndrome.
Disruption of complex I, also called NADH:ubiquinone oxidoreductase, is the most common cause of Leigh syndrome, accounting for nearly one third of cases of the condition. At least 25 genes involved in the formation of complex I, found in either nuclear or mitochondrial DNA, have been associated with Leigh syndrome.
Disruption of complex IV, also called cytochrome c oxidase or COX, is also a common cause of Leigh syndrome, underlying approximately 15 percent of cases. One of the most frequently altered genes in Leigh syndrome is SURF1. This gene, which is found in nuclear DNA, provides instructions for making a protein that helps assemble the COX protein complex (complex IV). This complex, which is involved in the last step of electron transfer in oxidative phosphorylation, provides the energy that will be used in the next step of the process to generate ATP. Variants in the SURF1 gene typically lead to an abnormally short SURF1 protein that is broken down in cells, resulting in the absence of functional SURF1 protein. The loss of this protein reduces the formation of normal COX complexes, which impairs mitochondrial energy production.
The most common mtDNA variant in Leigh syndrome affects the MT-ATP6 gene, which provides instructions for making a piece of complex V, also known as the ATP synthase protein complex . Using the energy provided by the other protein complexes, the ATP synthase complex generates ATP. MT-ATP6 gene variants , found in approximately 10 percent of people with Leigh syndrome, block the generation of ATP. Other mtDNA variants associated with Leigh syndrome decrease the activity of other oxidative phosphorylation protein complexes or lead to reduced formation of mitochondrial proteins, all of which impair mitochondrial energy production.
. Using the energy provided by the other protein complexes, the ATP synthase complex generates ATP. MT-ATP6 gene variants , found in approximately 10 percent of people with Leigh syndrome, block the generation of ATP. Other mtDNA variants associated with Leigh syndrome decrease the activity of other oxidative phosphorylation protein complexes or lead to reduced formation of mitochondrial proteins, all of which impair mitochondrial energy production.
Other gene variants associated with Leigh syndrome decrease the activity of one or more oxidative phosphorylation protein complexes or affect additional steps related to energy production. For example, Leigh syndrome can be caused by variants in genes that form the pyruvate dehydrogenase complex or coenzyme Q10, both of which are involved in mitochondrial energy production. Variants in genes that direct the replication of mtDNA or the production of mitochondrial proteins can also disrupt mitochondrial energy production.
Although the exact mechanism is unclear, researchers believe that impaired oxidative phosphorylation can lead to cell death because of decreased energy available in the cell. Certain tissues that require large amounts of energy, such as the brain, muscles, and heart, seem especially sensitive to decreases in cellular energy. Cell death in the brain likely causes the characteristic lesions seen in Leigh syndrome, which contribute to the signs and symptoms of the condition. Cell death in other sensitive tissues may also contribute to the features of Leigh syndrome.
Inheritance
Leigh syndrome can have different inheritance patterns. It is most commonly inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. This pattern of inheritance applies to most of the Leigh syndrome-associated genes contained in nuclear DNA, including SURF1. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. This pattern of inheritance applies to most of the Leigh syndrome-associated genes contained in nuclear DNA, including SURF1. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
In approximately 20 percent of people with Leigh syndrome, the condition is inherited in a mitochondrial pattern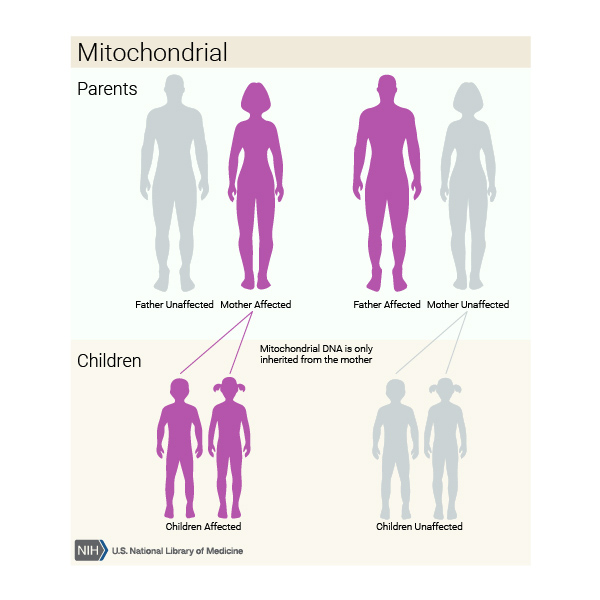 . This pattern of inheritance applies to genes contained in mtDNA, including MT-ATP6. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA variants only from their mother. Fathers do not pass traits associated with changes in mtDNA to their children, but when inherited from the mother, these disorders can appear in every generation of a family and can affect both males and females. Each cell has multiple copies of mtDNA. A variant is usually found in only some copies of mtDNA (known as heteroplasmy). The level of heteroplasmy can affect the severity of the condition. In some instances, a variant is found in all copies of mtDNA (known as homoplasmy).
. This pattern of inheritance applies to genes contained in mtDNA, including MT-ATP6. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA variants only from their mother. Fathers do not pass traits associated with changes in mtDNA to their children, but when inherited from the mother, these disorders can appear in every generation of a family and can affect both males and females. Each cell has multiple copies of mtDNA. A variant is usually found in only some copies of mtDNA (known as heteroplasmy). The level of heteroplasmy can affect the severity of the condition. In some instances, a variant is found in all copies of mtDNA (known as homoplasmy).
In a small number of affected individuals with variants in nuclear DNA, Leigh syndrome is inherited in an X-linked recessive pattern . The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes
. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes . In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a variant would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a variant would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Occasionally, Leigh syndrome is caused by genetic variants that occur spontaneously, and there is no family history of this condition.
Other Names for This Condition
- Infantile subacute necrotizing encephalopathy
- Juvenile subacute necrotizing encephalopathy
- Leigh disease
- Leigh's disease
- Subacute necrotizing encephalomyelopathy
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ball M, Thorburn DR, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome Spectrum. 2003 Oct 30 [updated 2024 May 9]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1173/ Citation on PubMed
- Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol. 2016 Feb;79(2):190-203. doi: 10.1002/ana.24551. Epub 2015 Dec 15. Citation on PubMed
- Pecina P, Capkova M, Chowdhury SK, Drahota Z, Dubot A, Vojtiskova A, Hansikova H, Houst'kova H, Zeman J, Godinot C, Houstek J. Functional alteration of cytochrome c oxidase by SURF1 mutations in Leigh syndrome. Biochim Biophys Acta. 2003 Sep 1;1639(1):53-63. doi: 10.1016/s0925-4439(03)00127-3. Citation on PubMed
- Rahman S, Thorburn DR, Ball M. Nuclear Gene-Encoded Leigh Syndrome Spectrum Overview. 2015 Oct 1 [updated 2025 May 1]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK320989/ Citation on PubMed
- Rahman S. Leigh syndrome. Handb Clin Neurol. 2023;194:43-63. doi: 10.1016/B978-0-12-821751-1.00015-4. Citation on PubMed
- Schubert Baldo M, Vilarinho L. Molecular basis of Leigh syndrome: a current look. Orphanet J Rare Dis. 2020 Jan 29;15(1):31. doi: 10.1186/s13023-020-1297-9. Citation on PubMed
- Sgarbi G, Baracca A, Lenaz G, Valentino LM, Carelli V, Solaini G. Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem J. 2006 May 1;395(3):493-500. doi: 10.1042/BJ20051748. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.