

Description
Mitochondrial complex I deficiency is a shortage (deficiency) of a protein complex called complex I or a loss of its function. Complex I is found in cell structures called mitochondria, which convert the energy from food into a form that cells can use. Complex I is the first of five mitochondrial complexes that carry out a multi-step process called oxidative phosphorylation, through which cells derive much of their energy.

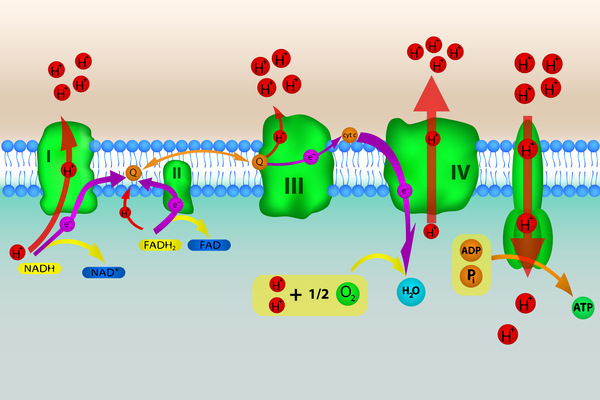
Mitochondrial complex I deficiency can cause a wide variety of signs and symptoms affecting many organs and systems of the body, particularly the nervous system, the heart, and the muscles used for movement (skeletal muscles). These signs and symptoms can appear at any time from birth to adulthood.
People with mitochondrial complex I deficiency typically have neurological problems, such as abnormal brain function (encephalopathy), recurrent seizures (epilepsy), intellectual disability, difficulty coordinating movements (ataxia), or involuntary movements (dystonia). Affected individuals may have low muscle tone (hypotonia), muscle pain (myalgia), and extreme fatigue in response to physical activity (exercise intolerance). They tend to develop elevated levels of lactic acid in the blood (lactic acidosis), which can cause nausea, vomiting, weakness, and rapid breathing. In severe cases, lactic acidosis can be life-threatening.
People with mitochondrial complex I deficiency sometimes have heart, liver, or kidney problems. Vision problems due to abnormal eye movement or breakdown (degeneration) of the nerves that carry signals from the eyes to the brain (optic nerves) can also occur.
Some people with mitochondrial complex I deficiency have groups of signs and symptoms that are classified as a specific syndrome. For example, a condition called Leigh syndrome is most commonly caused by mitochondrial complex I deficiency. Leigh syndrome is characterized by progressive loss of mental and movement abilities (developmental or psychomotor regression) and typically results in death within 2 to 3 years from the onset of symptoms. Another condition that can be caused by mitochondrial complex I deficiency, Leber hereditary optic neuropathy, is associated mainly with vision problems due to optic nerve degeneration. These syndromes can also have other causes.
Frequency
Mitochondrial diseases are thought to occur in about 1 in 8,500 people. Mitochondrial complex I deficiency is the most common cause of mitochondrial disease in children, accounting for approximately 30 percent of cases.
Causes
Mutations in many genes can cause mitochondrial complex I deficiency. Most of these genes provide instructions for making components of complex I or proteins that help assemble the complex. In some cases, the genes are involved in other functions that influence these processes.
Mutations that cause mitochondrial complex I deficiency impair the formation or function of complex I. As a result, complex I activity is reduced and oxidative phosphorylation is impaired. Researchers believe that problems with oxidative phosphorylation can lead to cell death by reducing the amount of energy available in the cell. It is thought that tissues and organs that require a lot of energy, such as the nervous system, heart, liver, kidneys, and skeletal muscles, are most affected by a reduction in oxidative phosphorylation.
Most genes known to be involved in mitochondrial complex I deficiency are found in nuclear DNA, which is packaged in chromosomes within the cell nucleus. Other genes involved in the condition are found in mitochondrial DNA (mtDNA), which is located in the mitochondria themselves. Most of the body's cells contain many mitochondria, and the mitochondria each contain many sets of mtDNA. When a mutation occurs in mtDNA, either all the mtDNA will have the same change (homoplasmy), or just some of the mtDNA will contain the change (heteroplasmy). A higher percentage of mutated mtDNA typically causes more severe disease.
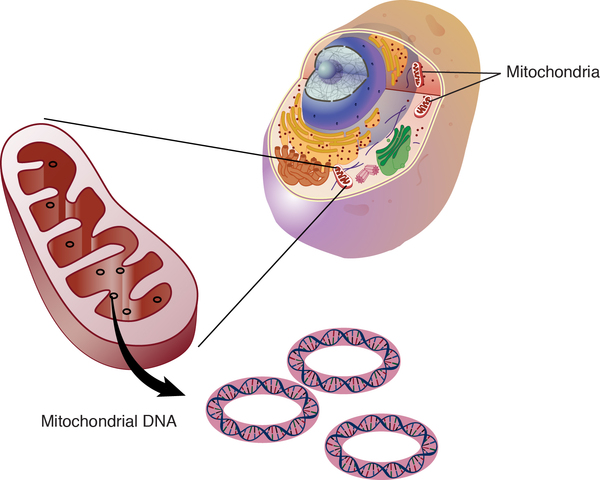
Inheritance
Mitochondrial complex I deficiency has several inheritance patterns, depending on the gene involved. When the disorder is caused by a mutation in a gene found in nuclear DNA, it has autosomal recessive or X-linked inheritance. Autosomal recessive means that both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition because the other copy of the gene is normal.

X-linked inheritance occurs when the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males, who have only one X chromosome, a mutation in the only copy of the gene in each cell is sufficient to cause the condition. In females, who have two copies of the X chromosome, one altered copy of the gene in each cell can lead to less severe features of the condition or may cause no signs or symptoms at all. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

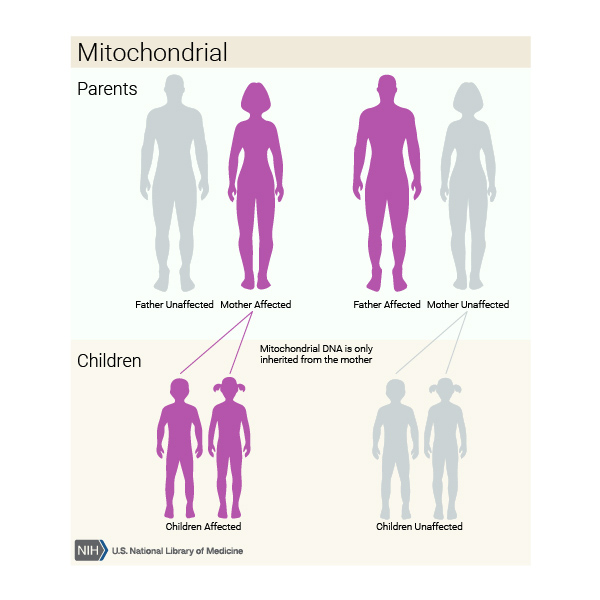
When mitochondrial complex I deficiency is caused by a mutation in a gene found in mtDNA, it is inherited in a mitochondrial pattern, which is also known as maternal inheritance. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA mutations only from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
Other Names for This Condition
- NADH-coenzyme Q reductase deficiency
- NADH:Q(1) oxidoreductase deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017 Jan;241(2):236-250. doi: 10.1002/path.4809. Epub 2016 Nov 2. Citation on PubMed or Free article on PubMed Central
- Blanchet L, Buydens MC, Smeitink JA, Willems PH, Koopman WJ. Isolated mitochondrial complex I deficiency: explorative data analysis of patient cell parameters. Curr Pharm Des. 2011 Dec 1;17(36):4023-33. doi: 10.2174/138161211798764870. Citation on PubMed
- Distelmaier F, Koopman WJ, van den Heuvel LP, Rodenburg RJ, Mayatepek E, Willems PH, Smeitink JA. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009 Apr;132(Pt 4):833-42. doi: 10.1093/brain/awp058. Epub 2009 Mar 31. Citation on PubMed
- Fernandez-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta. 2009 Jan;1793(1):200-11. doi: 10.1016/j.bbamcr.2008.05.028. Epub 2008 Jun 21. Citation on PubMed
- Leman G, Gueguen N, Desquiret-Dumas V, Kane MS, Wettervald C, Chupin S, Chevrollier A, Lebre AS, Bonnefont JP, Barth M, Amati-Bonneau P, Verny C, Henrion D, Bonneau D, Reynier P, Procaccio V. Assembly defects induce oxidative stress in inherited mitochondrial complex I deficiency. Int J Biochem Cell Biol. 2015 Aug;65:91-103. doi: 10.1016/j.biocel.2015.05.017. Epub 2015 May 27. Citation on PubMed
- Pagniez-Mammeri H, Loublier S, Legrand A, Benit P, Rustin P, Slama A. Mitochondrial complex I deficiency of nuclear origin I. Structural genes. Mol Genet Metab. 2012 Feb;105(2):163-72. doi: 10.1016/j.ymgme.2011.11.188. Epub 2011 Nov 18. Citation on PubMed
- Pagniez-Mammeri H, Rak M, Legrand A, Benit P, Rustin P, Slama A. Mitochondrial complex I deficiency of nuclear origin II. Non-structural genes. Mol Genet Metab. 2012 Feb;105(2):173-9. doi: 10.1016/j.ymgme.2011.10.001. Epub 2011 Oct 20. Citation on PubMed
- Vartak RS, Semwal MK, Bai Y. An update on complex I assembly: the assembly of players. J Bioenerg Biomembr. 2014 Aug;46(4):323-8. doi: 10.1007/s10863-014-9564-x. Epub 2014 Jul 17. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.