

Description
Miller syndrome is a rare condition that mainly affects the development of the face and limbs. The severity of this disorder varies among affected individuals.
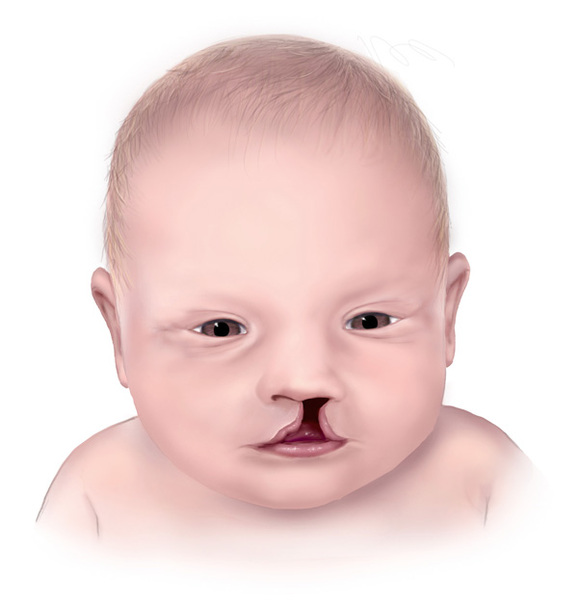
Children with Miller syndrome are born with underdeveloped cheek bones (malar hypoplasia) and a very small lower jaw (micrognathia). They often have an opening in the roof of the mouth (cleft palate) and/or a split in the upper lip (cleft lip). These abnormalities frequently cause feeding problems in infants with Miller syndrome. The airway is usually restricted due to the micrognathia, which can lead to life-threatening breathing problems.
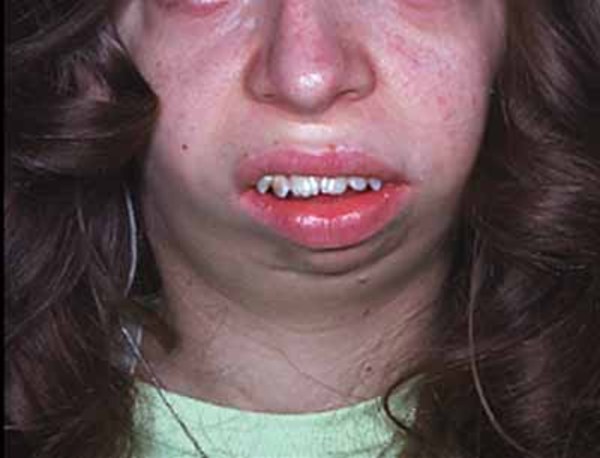

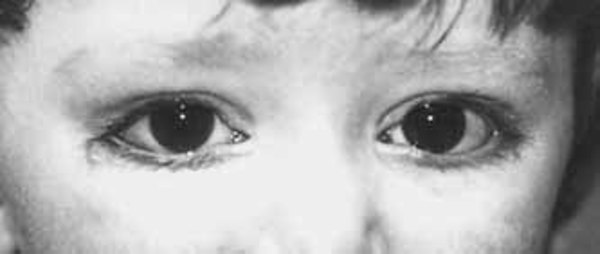
People with Miller syndrome often have eyes that slant downward, eyelids that turn out so the inner surface is exposed (ectropion), and a notch in the lower eyelids called an eyelid coloboma. Many affected individuals have small, cup-shaped ears, and some have hearing loss caused by defects in the middle ear (conductive hearing loss). Another feature of this condition is the presence of extra nipples. Miller syndrome does not affect a person's intelligence, although speech development may be delayed due to hearing impairment.



Individuals with Miller syndrome have various bone abnormalities in their arms and legs. The most common problem is absent fifth (pinky) fingers and toes. Affected individuals may also have webbed or fused fingers or toes (syndactyly) and abnormally formed bones in the forearms and lower legs. People with Miller syndrome sometimes have defects in other bones, such as the ribs or spine.
Less commonly, affected individuals have abnormalities of the heart, kidneys, genitalia, or gastrointestinal tract.
Frequency
Miller syndrome is a rare disorder; it is estimated to affect fewer than 1 in 1 million newborns. At least 30 cases have been reported in the medical literature.
Causes
Mutations in the DHODH gene cause Miller syndrome. This gene provides instructions for making an enzyme called dihydroorotate dehydrogenase. This enzyme is involved in producing pyrimidines, which are building blocks of DNA, its chemical cousin RNA, and molecules such as ATP and GTP that serve as energy sources in the cell. Specifically, dihydroorotate dehydrogenase converts a molecule called dihydroorotate to a molecule called orotic acid. In subsequent steps, other enzymes modify orotic acid to produce pyrimidines.
Miller syndrome disrupts the development of structures called the first and second pharyngeal arches. The pharyngeal arches are five paired structures that form on each side of the head and neck during embryonic development. These structures develop into the bones, skin, nerves, and muscles of the head and neck. In particular, the first and second pharyngeal arches develop into the jaw, the nerves and muscles for chewing and facial expressions, the bones in the middle ear, and the outer ear. It remains unclear exactly how DHODH gene mutations lead to abnormal development of the pharyngeal arches in people with Miller syndrome.
Development of the arms and legs is also affected by Miller syndrome. Each limb starts out as a small mound of tissue called a limb bud, which grows outward. Many different proteins are involved in the normal shaping (patterning) of each limb. Once the overall pattern of a limb is formed, detailed shaping can take place. For example, to create individual fingers and toes, certain cells self-destruct (undergo apoptosis) to remove the webbing between each digit. The role dihydroorotate dehydrogenase plays in limb development is not known. It is also unknown how mutations in the DHODH gene cause bone abnormalities in the arms and legs of people with Miller syndrome.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Genee-Wiedemann acrofacial dysostosis
- Genee-Wiedemann syndrome
- Postaxial acrofacial dysostosis (POADS)
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Biesecker LG. Exome sequencing makes medical genomics a reality. Nat Genet. 2010 Jan;42(1):13-4. doi: 10.1038/ng0110-13. Citation on PubMed
- Brosnan ME, Brosnan JT. Orotic acid excretion and arginine metabolism. J Nutr. 2007 Jun;137(6 Suppl 2):1656S-1661S. doi: 10.1093/jn/137.6.1656S. Citation on PubMed
- Gurrieri F, Kjaer KW, Sangiorgi E, Neri G. Limb anomalies: Developmental and evolutionary aspects. Am J Med Genet. 2002 Dec 30;115(4):231-44. doi: 10.1002/ajmg.10981. Citation on PubMed
- Neumann L, Pelz J, Kunze J. A new observation of two cases of acrofacial dysostosis type Genee-Wiedemann in a family--remarks on the mode of inheritance: report on two sibs. Am J Med Genet. 1996 Sep 6;64(4):556-62. doi: 10.1002/(SICI)1096-8628(19960906)64:43.0.CO;2-N. Citation on PubMed
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010 Jan;42(1):30-5. doi: 10.1038/ng.499. Epub 2009 Nov 13. Citation on PubMed or Free article on PubMed Central
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010 Apr 30;328(5978):636-9. doi: 10.1126/science.1186802. Epub 2010 Mar 10. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.