

Description
McKusick-Kaufman syndrome is a condition that affects the development of the hands, feet, heart, and reproductive system. It is characterized by a combination of three features: extra fingers and/or toes (polydactyly), congenital heart defects, and genital abnormalities. The most common genital abnormality is hydrometrocolpos, an accumulation of fluid in the vagina and uterus.
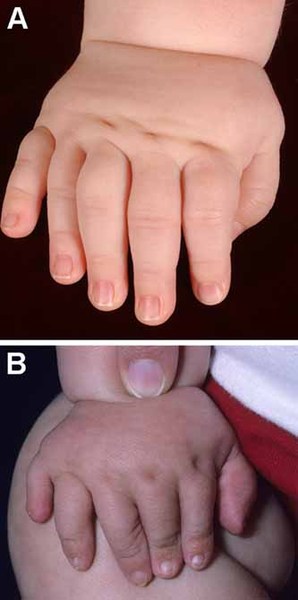
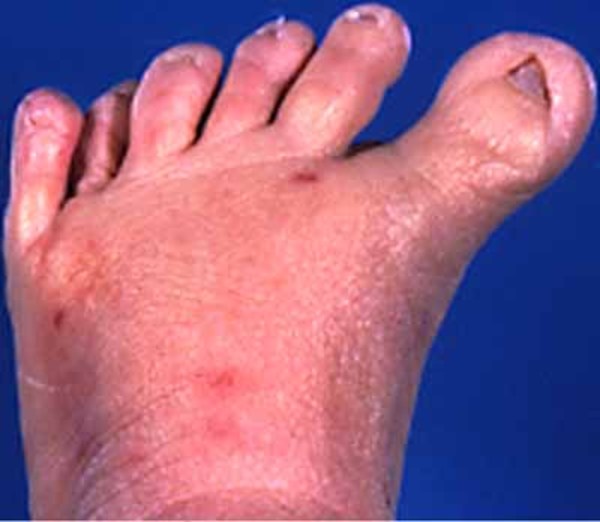
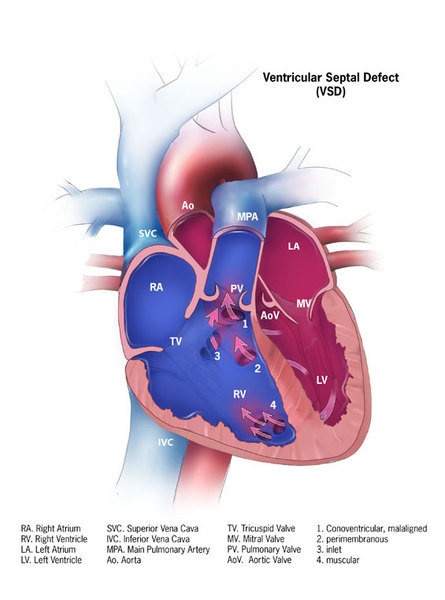
In people with McKusick-Kaufman syndrome, the extra digits are typically on the same side of the hand or foot as the pinky or little toe (postaxial polydactyly). The congenital heart defects in individuals with McKusick-Kaufman syndrome can include an atrial septal defect or a ventricular septal defect, which are openings in the wall (septum) that separates the upper or lower chambers of the heart.

A genital abnormality called hydrometrocolpos is common in individuals with McKusick-Kaufman syndrome. Hydrometrocolpos can occur if part of the vagina fails to develop (vaginal agenesis) or if a membrane blocks the opening of the vagina. The blockage allows fluid to build up in the vagina and uterus, stretching these organs and leading to a fluid-filled mass. Other genital abnormalities associated with McKusick-Kaufman syndrome can include a urethral opening on the underside of the penis (hypospadias), a downward-curving penis (chordee), and undescended testes (cryptorchidism).
The signs and symptoms of McKusick-Kaufman syndrome overlap significantly with those of another genetic disorder, Bardet-Biedl syndrome. However, Bardet-Biedl syndrome has several features that are not typically seen in people with McKusick-Kaufman syndrome. These include a gradual loss of vision, developmental disabilities, kidney abnormalities, and obesity. Because some of these features are not apparent at birth, the two conditions can be difficult to tell apart in infancy and early childhood.
Both McKusick-Kaufman syndrome and Bardet-Biedl syndrome belong to a group of conditions called ciliopathies. Ciliopathies are inherited disorders that affect the structure or function of cilia, the microscopic, finger-like projections found on the surface of cells. Cilia are involved in signaling pathways that transmit information between cells.
Frequency
More than 100 individuals with McKusick-Kaufman syndrome have been reported in the literature. This condition was first described in the Old Order Amish population, where it affects an estimated 1 in 10,000 people.
The incidence of McKusick-Kaufman syndrome in non-Amish populations is unknown.
Causes
Some variants (also called mutations) in the MKKS gene have been found to cause McKusick-Kaufman syndrome. The MKKS gene is also called the BBS6 gene. This gene provides instructions for making a protein that plays an important role in early development, specifically in the formation of the limbs, heart, and reproductive system. The protein's structure suggests that it may belong to a family of proteins called chaperonins. Proteins must be folded into the correct shape to function properly, and chaperonins help them do that.

The MKKS protein is thought to play a role in cell division and cell transport. Specifically, the MKKS protein is thought to transport molecules between the cytoplasm and the nucleus of the cell. The MKKS protein also combines with other proteins to form a complex within the cell. This complex is used to assemble the molecules involved in transporting materials that support the function of cilia.
Though it is not clear exactly how variants in the MKKS gene lead to the specific signs and symptoms of McKusick-Kaufman syndrome, two particular variants in the MKKS gene appear to impair the protein's ability to transport a molecule involved in regulating gene activity. This change likely affects the activity of certain genes that are critical during early development.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant that causes the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant that causes the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- HMCS
- Hydrometrocolpos syndrome
- Hydrometrocolpos, postaxial polydactyly, and congenital heart malformation
- Hydrometrocolpos-postaxial polydactyly syndrome
- Kaufman-McKusick syndrome
- MKS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- David A, Bitoun P, Lacombe D, Lambert JC, Nivelon A, Vigneron J, Verloes A. Hydrometrocolpos and polydactyly: a common neonatal presentation of Bardet-Biedl and McKusick-Kaufman syndromes. J Med Genet. 1999 Aug;36(8):599-603. Citation on PubMed or Free article on PubMed Central
- Scott CA, Marsden AN, Rebagliati MR, Zhang Q, Chamling X, Searby CC, Baye LM, Sheffield VC, Slusarski DC. Nuclear/cytoplasmic transport defects in BBS6 underlie congenital heart disease through perturbation of a chromatin remodeling protein. PLoS Genet. 2017 Jul 28;13(7):e1006936. doi: 10.1371/journal.pgen.1006936. eCollection 2017 Jul. Citation on PubMed
- Seo S, Baye LM, Schulz NP, Beck JS, Zhang Q, Slusarski DC, Sheffield VC. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci U S A. 2010 Jan 26;107(4):1488-93. doi: 10.1073/pnas.0910268107. Epub 2010 Jan 4. Citation on PubMed or Free article on PubMed Central
- Slavotinek AM, Biesecker LG. Phenotypic overlap of McKusick-Kaufman syndrome with bardet-biedl syndrome: a literature review. Am J Med Genet. 2000 Nov 27;95(3):208-15. Citation on PubMed
- Slavotinek AM. McKusick-Kaufman Syndrome. 2002 Sep 10 [updated 2020 Dec 3]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1502/ Citation on PubMed
- Stone DL, Slavotinek A, Bouffard GG, Banerjee-Basu S, Baxevanis AD, Barr M, Biesecker LG. Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome. Nat Genet. 2000 May;25(1):79-82. doi: 10.1038/75637. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.