

Description
MBD5-associated neurodevelopmental disorder (MAND) is a condition that affects neurological and physical development.
Children with MAND have mild to severe intellectual disability and developmental delay. They often have poor coordination and do not walk until age 2 or 3. Their walking style (gait) is often unbalanced and wide-based. Language skills, both the production of speech and the ability to understand speech, are very limited in affected individuals. By age 2, most children with MAND develop recurring seizures (epilepsy). Most affected children have feeding problems due to weak muscle tone (hypotonia). Constipation also frequently occurs.
Sleep problems are common in MAND and include night terrors, waking frequently during the night, and waking early in the morning. As a result, many affected individuals are extremely tired during the day due to lack of sleep and poor-quality sleep. Most people with MAND have features similar to autism spectrum disorder, a developmental condition that affects communication and social interaction. They have a short attention span; perform repetitive hand movements (stereotypies), such as clapping, hand licking, and hand sucking; and grind their teeth.
People with MAND tend to have subtle facial features, including a broad forehead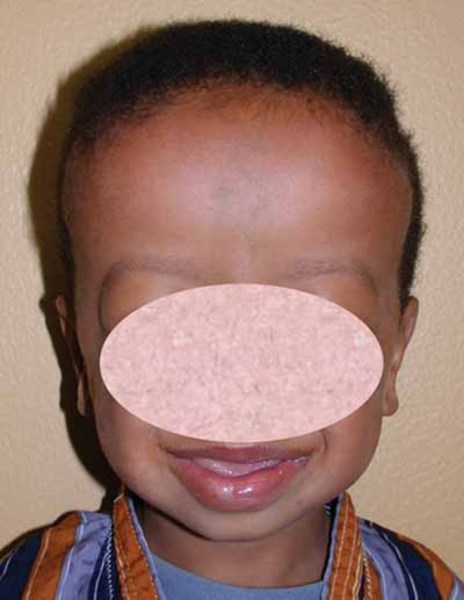 , thick and highly arched eyebrows, abnormalities of the outer ear, a short nose
, thick and highly arched eyebrows, abnormalities of the outer ear, a short nose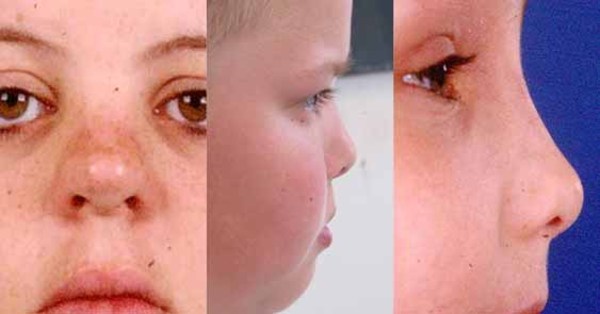 , a wide
, a wide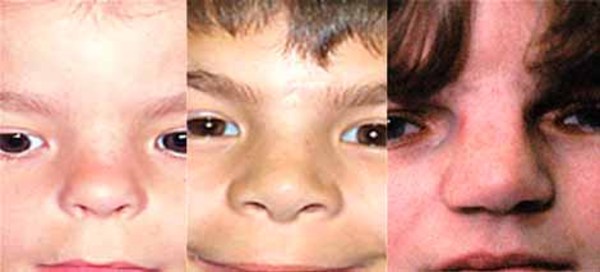 or depressed nasal bridge
or depressed nasal bridge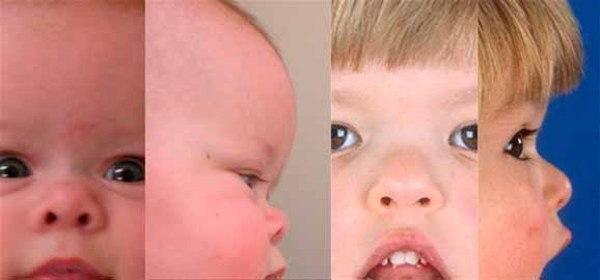 , downturned corners of the mouth, an upper lip that points outward (called a tented lip), and a full lower lip. Some affected individuals have mild skeletal abnormalities including small hands
, downturned corners of the mouth, an upper lip that points outward (called a tented lip), and a full lower lip. Some affected individuals have mild skeletal abnormalities including small hands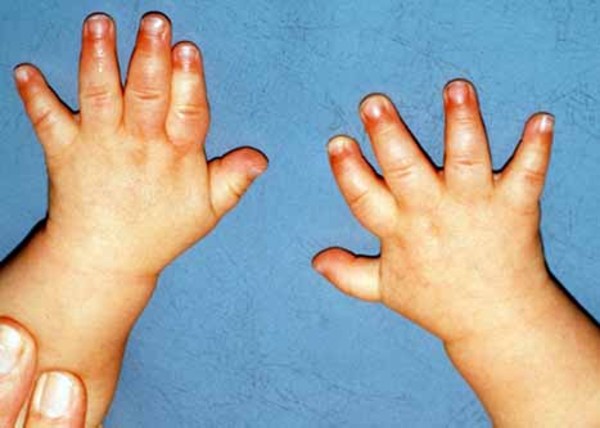 and feet, short fingers
and feet, short fingers (brachydactyly), curved pinky fingers (fifth-finger clinodactyly
(brachydactyly), curved pinky fingers (fifth-finger clinodactyly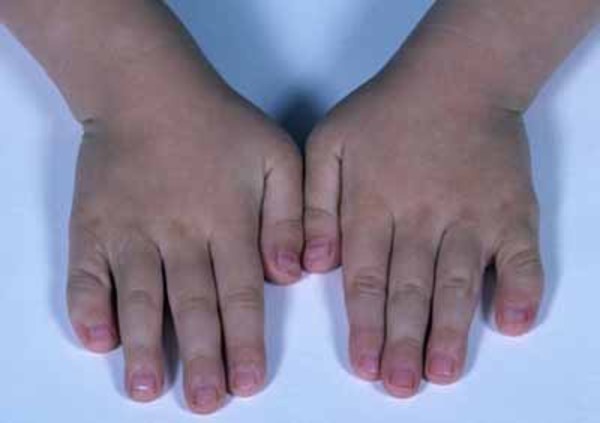 ), or a wide gap between the first and second toes (known as a sandal gap
), or a wide gap between the first and second toes (known as a sandal gap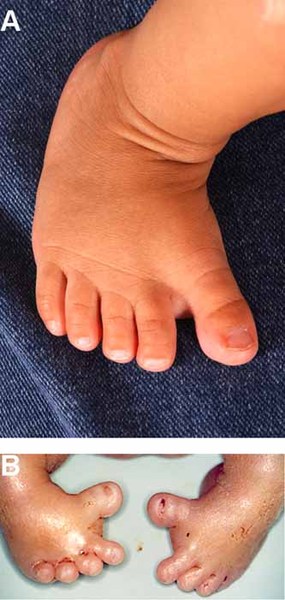 ). Rarely, individuals with MAND have heart abnormalities.
). Rarely, individuals with MAND have heart abnormalities.
Frequency
MAND is thought to be a rare disorder, although its prevalence is unknown. More than 100 affected individuals have been described in the scientific literature.
Causes
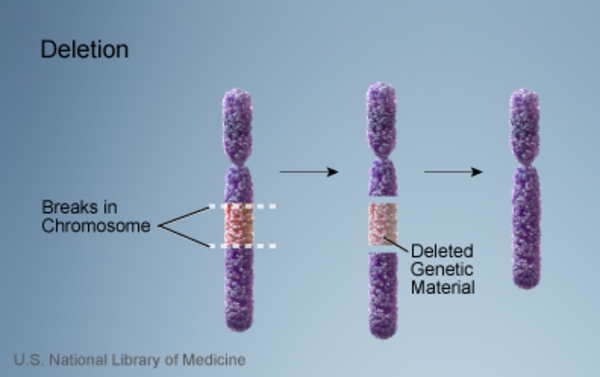
MAND is most often caused by a loss (deletion) or gain (duplication) of genetic material in a particular region of chromosome 2. These changes are also known either as 2q23.1 microdeletions or 2q23.1 microduplications. The missing or extra region varies in length from a few thousand to a few million DNA building blocks (base pairs) but always includes the MBD5 gene. Less frequently, MAND is caused by mutations that affect only the MBD5 gene. Because mutations in the MBD5 gene and changes on chromosome 2 that involve this gene both cause MAND, researchers believe that MBD5 gene changes underlie most of the signs and symptoms of the condition. Neurological features of the condition generally do not differ based on the genetic cause, although they can vary between individuals.
The MBD5 gene provides instructions for a protein that likely regulates the activity (expression) of genes, controlling the production of proteins that are involved in neurological functions such as learning, memory, and behavior. The MBD5 protein also seems to play a role in the growth and division (proliferation) and maturation (differentiation) of various types of cells.
All of the genetic changes associated with MAND lead to an abnormal amount of MBD5 protein. MBD5 gene mutations and deletions prevent one copy of the MBD5 gene in each cell from producing any functional protein, which reduces the total amount of this protein in cells. A duplication leads to an increased amount of MBD5 protein. It is likely that any changes in MBD5 protein levels impair its regulation of gene expression, leading to the uncontrolled production of certain proteins. Proteins that play a role in neurological functions are particularly affected, which helps explain why MAND impacts brain development and behavior. An increase or decrease in MBD5 protein disrupts gene expression that is normally well-controlled by this protein, which is likely why duplications and deletions involving the MBD5 gene lead to the same signs and symptoms. The cause of the skeletal abnormalities and other non-neurological features of MAND is unclear. It is also unknown whether the loss or gain of other genes in chromosome 2 deletions or duplications contributes to the variable features of MAND.
Inheritance
MAND is considered an autosomal dominant condition because one copy of the altered chromosome 2 or MBD5 gene in each cell is sufficient to cause the disorder. Most cases of MAND are not inherited but occur as random events during the formation of reproductive cells (eggs or sperm) in a parent of an affected individual. These cases occur in people with no history of the disorder in their family. In a small percentage of cases, people with MAND inherit the altered chromosome or gene from a parent with the condition.


Other Names for This Condition
- 2q23.1 microdeletion syndrome
- 2q23.1 microduplication syndrome
- MAND
- MBD5 haploinsufficiency
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Hodge JC, Mitchell E, Pillalamarri V, Toler TL, Bartel F, Kearney HM, Zou YS, Tan WH, Hanscom C, Kirmani S, Hanson RR, Skinner SA, Rogers RC, Everman DB, Boyd E, Tapp C, Mullegama SV, Keelean-Fuller D, Powell CM, Elsea SH, Morton CC, Gusella JF, DuPont B, Chaubey A, Lin AE, Talkowski ME. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry. 2014 Mar;19(3):368-79. doi: 10.1038/mp.2013.42. Epub 2013 Apr 16. Citation on PubMed or Free article on PubMed Central
- Mullegama SV, Elsea SH. Clinical and Molecular Aspects of MBD5-Associated Neurodevelopmental Disorder (MAND). Eur J Hum Genet. 2016 Aug;24(9):1235-43. doi: 10.1038/ejhg.2016.35. Epub 2016 May 25. Citation on PubMed or Free article on PubMed Central
- Mullegama SV, Pugliesi L, Burns B, Shah Z, Tahir R, Gu Y, Nelson DL, Elsea SH. MBD5 haploinsufficiency is associated with sleep disturbance and disrupts circadian pathways common to Smith-Magenis and fragile X syndromes. Eur J Hum Genet. 2015 Jun;23(6):781-9. doi: 10.1038/ejhg.2014.200. Epub 2014 Oct 1. Citation on PubMed or Free article on PubMed Central
- Mullegama SV, Rosenfeld JA, Orellana C, van Bon BW, Halbach S, Repnikova EA, Brick L, Li C, Dupuis L, Rosello M, Aradhya S, Stavropoulos DJ, Manickam K, Mitchell E, Hodge JC, Talkowski ME, Gusella JF, Keller K, Zonana J, Schwartz S, Pyatt RE, Waggoner DJ, Shaffer LG, Lin AE, de Vries BB, Mendoza-Londono R, Elsea SH. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet. 2014 Jan;22(1):57-63. doi: 10.1038/ejhg.2013.67. Epub 2013 May 1. Citation on PubMed or Free article on PubMed Central
- Tadros S, Wang R, Waters JJ, Waterman C, Collins AL, Collinson MN, Ahn JW, Josifova D, Chetan R, Kumar A. Inherited 2q23.1 microdeletions involving the MBD5 locus. Mol Genet Genomic Med. 2017 Aug 8;5(5):608-613. doi: 10.1002/mgg3.316. eCollection 2017 Sep. Citation on PubMed or Free article on PubMed Central
- Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, Gastier-Foster J, Thrush DL, Kathiresan S, Ruderfer DM, Chiang C, Hanscom C, Ernst C, Lindgren AM, Morton CC, An Y, Astbury C, Brueton LA, Lichtenbelt KD, Ades LC, Fichera M, Romano C, Innis JW, Williams CA, Bartholomew D, Van Allen MI, Parikh A, Zhang L, Wu BL, Pyatt RE, Schwartz S, Shaffer LG, de Vries BB, Gusella JF, Elsea SH. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011 Oct 7;89(4):551-63. doi: 10.1016/j.ajhg.2011.09.011. Citation on PubMed or Free article on PubMed Central
- Walz K, Young JI. The methyl binding domain containing protein MBD5 is a transcriptional regulator responsible for 2q23.1 deletion syndrome. Rare Dis. 2014 Nov 3;2(1):e967151. doi: 10.4161/2167549X.2014.967151. eCollection 2014. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.