

Description
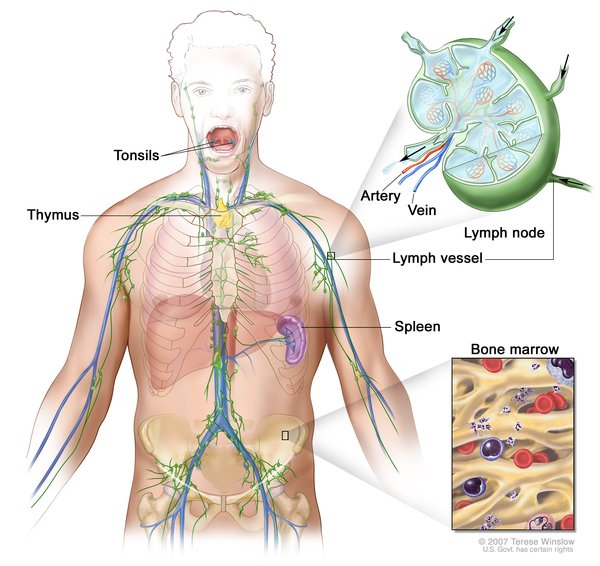
Lymphangioleiomyomatosis (LAM) is a condition that affects the lungs, the kidneys, and the lymphatic system. The lymphatic system consists of a network of vessels that transport lymph fluid and immune cells throughout the body. Lymph fluid helps exchange immune cells, proteins, and other substances between the blood and tissues.
LAM is found almost exclusively in women. It often occurs as a feature of an inherited syndrome called tuberous sclerosis complex. When LAM occurs alone it is called isolated or sporadic LAM.
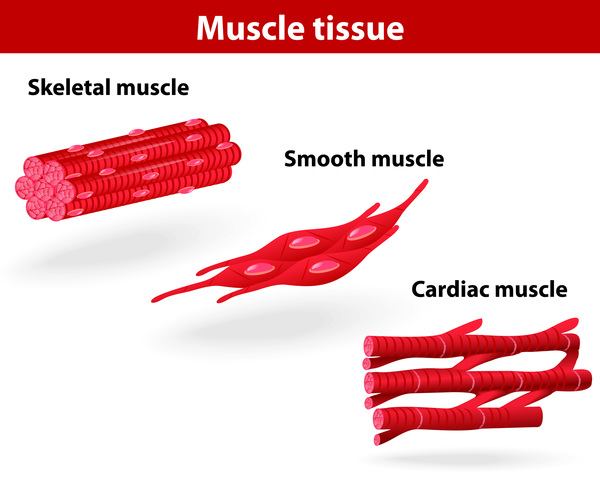
Signs and symptoms of LAM most often appear during a woman's thirties. Affected women have an overgrowth of abnormal smooth muscle-like cells (LAM cells) in the lungs, resulting in the formation of lung cysts and the destruction of normal lung tissue. They may also have an accumulation of fluid in the cavity around the lungs (chylothorax).
The lung abnormalities resulting from LAM may cause difficulty breathing (dyspnea), chest pain, and coughing, which may bring up blood (hemoptysis). Many women with this disorder have recurrent episodes of collapsed lung (spontaneous pneumothorax). The lung problems may be progressive and, without lung transplantation, may eventually lead to limitations in activities of daily living, the need for oxygen therapy, and respiratory failure. Although LAM cells are not considered cancerous, they may spread between tissues (metastasize). As a result, the condition may recur even after lung transplantation.
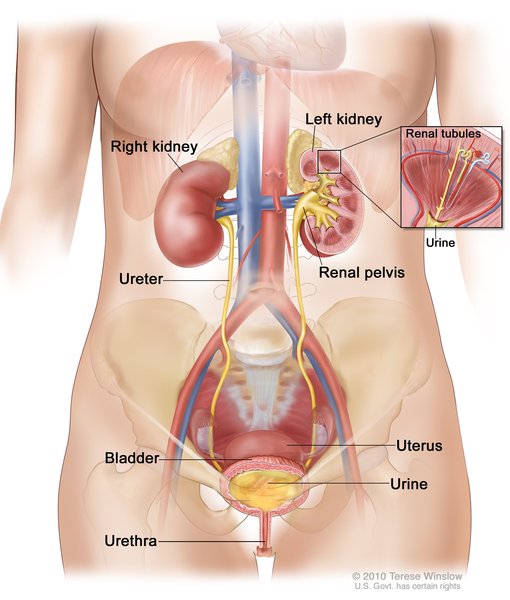
Women with LAM may develop cysts in the lymphatic vessels of the chest and abdomen. These cysts are called lymphangioleiomyomas. Affected women may also develop tumors called angiomyolipomas made up of LAM cells, fat cells, and blood vessels. Angiomyolipomas usually develop in the kidneys. Internal bleeding is a common complication of angiomyolipomas.
Frequency
LAM occurs in approximately 30 percent of women with tuberous sclerosis complex. Sporadic LAM, which occurs without tuberous sclerosis complex, is estimated to affect 3.3 to 7.4 per million women worldwide. This condition may be underdiagnosed because its symptoms are similar to those of other lung disorders such as asthma, bronchitis, and chronic obstructive pulmonary disease.
Causes
Mutations in the TSC1 gene or, more commonly, the TSC2 gene, cause LAM. The TSC1 and TSC2 genes provide instructions for making the proteins hamartin and tuberin, respectively. Within cells, these two proteins likely help regulate cell growth and size. The proteins act as tumor suppressors, which normally prevent cells from growing and dividing too fast or in an uncontrolled way.
When both copies of the TSC1 gene are mutated in a particular cell, that cell cannot produce any functional hamartin; cells with two altered copies of the TSC2 gene are unable to produce any functional tuberin. The loss of these proteins allows the cell to grow and divide in an uncontrolled way, resulting in the tumors and cysts associated with LAM.
It is not well understood why LAM occurs predominantly in women. Researchers believe that the female sex hormone estrogen may be involved in the development of the disorder.
Inheritance
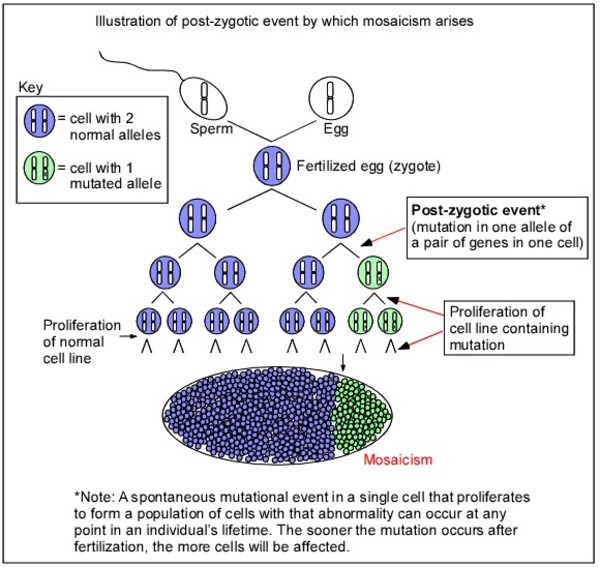
Sporadic LAM is not inherited. Instead, researchers suggest that it is caused by a random mutation in the TSC1 or TSC2 gene that occurs very early in development. As a result, some of the body's cells have a normal version of the gene, while others have the mutated version. This situation is called mosaicism. When a mutation occurs in the other copy of the TSC1 or TSC2 gene in certain cells during a woman's lifetime (a somatic mutation), she may develop LAM. These women typically have no history of this disorder in their family.
Other Names for This Condition
- LAM
- Lymphangiomyomatosis
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Harari S, Torre O, Cassandro R, Moss J. The changing face of a rare disease: lymphangioleiomyomatosis. Eur Respir J. 2015 Nov;46(5):1471-85. doi: 10.1183/13993003.00412-2015. Epub 2015 Sep 24. Citation on PubMed
- Harari S, Torre O, Moss J. Lymphangioleiomyomatosis: what do we know and what are we looking for? Eur Respir Rev. 2011 Mar;20(119):34-44. doi: 10.1183/09059180.00011010. Citation on PubMed or Free article on PubMed Central
- Henske EP, McCormack FX. Lymphangioleiomyomatosis - a wolf in sheep's clothing. J Clin Invest. 2012 Nov;122(11):3807-16. doi: 10.1172/JCI58709. Epub 2012 Nov 1. Citation on PubMed or Free article on PubMed Central
- Johnson SR, Taveira-DaSilva AM, Moss J. Lymphangioleiomyomatosis. Clin Chest Med. 2016 Sep;37(3):389-403. doi: 10.1016/j.ccm.2016.04.002. Citation on PubMed
- Martignoni G, Pea M, Reghellin D, Gobbo S, Zamboni G, Chilosi M, Bonetti F. Molecular pathology of lymphangioleiomyomatosis and other perivascular epithelioid cell tumors. Arch Pathol Lab Med. 2010 Jan;134(1):33-40. doi: 10.5858/2008-0542-RAR1.1. Citation on PubMed
- Meraj R, Wikenheiser-Brokamp KA, Young LR, McCormack FX. Lymphangioleiomyomatosis: new concepts in pathogenesis, diagnosis, and treatment. Semin Respir Crit Care Med. 2012 Oct;33(5):486-97. doi: 10.1055/s-0032-1325159. Epub 2012 Sep 21. Citation on PubMed
- Moir LM. Lymphangioleiomyomatosis: Current understanding and potential treatments. Pharmacol Ther. 2016 Feb;158:114-24. doi: 10.1016/j.pharmthera.2015.12.008. Epub 2015 Dec 20. Citation on PubMed
- Radzikowska E. Lymphangioleiomyomatosis: New Treatment Perspectives. Lung. 2015 Aug;193(4):467-75. doi: 10.1007/s00408-015-9742-6. Epub 2015 May 17. Citation on PubMed
- Taveira-DaSilva AM, Pacheco-Rodriguez G, Moss J. The natural history of lymphangioleiomyomatosis: markers of severity, rate of progression and prognosis. Lymphat Res Biol. 2010 Mar;8(1):9-19. doi: 10.1089/lrb.2009.0024. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.