

Description
Lowe syndrome is a condition that primarily affects the eyes, brain, and kidneys. This disorder occurs almost exclusively in males.
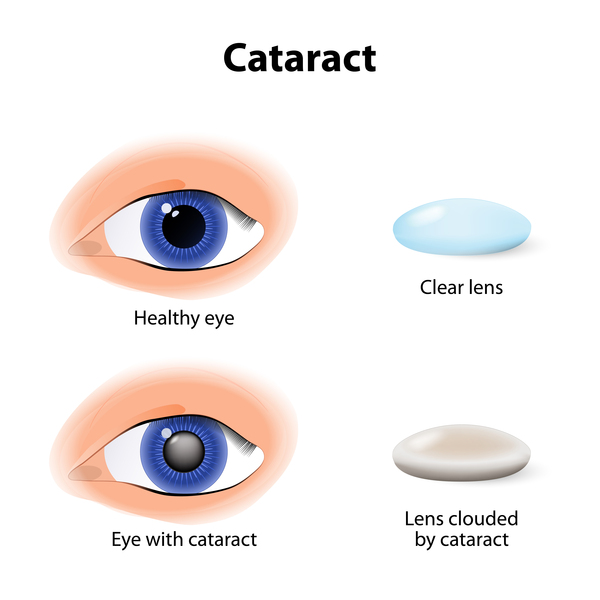
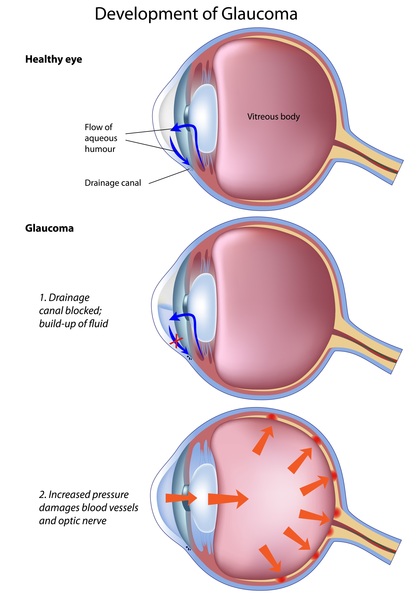
Infants with Lowe syndrome are born with thick clouding of the lenses in both eyes (congenital cataracts), often with other eye abnormalities that can impair vision. About half of affected infants develop an eye disease called infantile glaucoma, which is characterized by increased pressure within the eyes.
Many individuals with Lowe syndrome have delayed development, and intellectual ability ranges from normal to severely impaired. Behavioral problems and seizures have also been reported in children with this condition. Most affected children have weak muscle tone from birth (neonatal hypotonia), which can contribute to feeding difficulties, problems with breathing, and delayed development of motor skills such as sitting, standing, and walking.
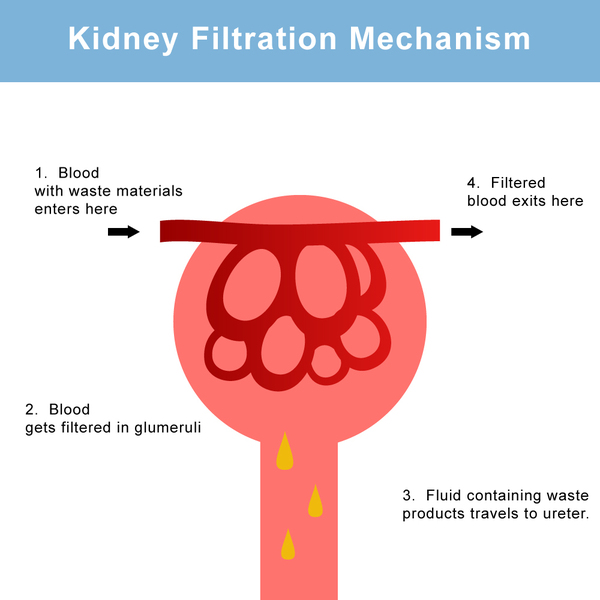
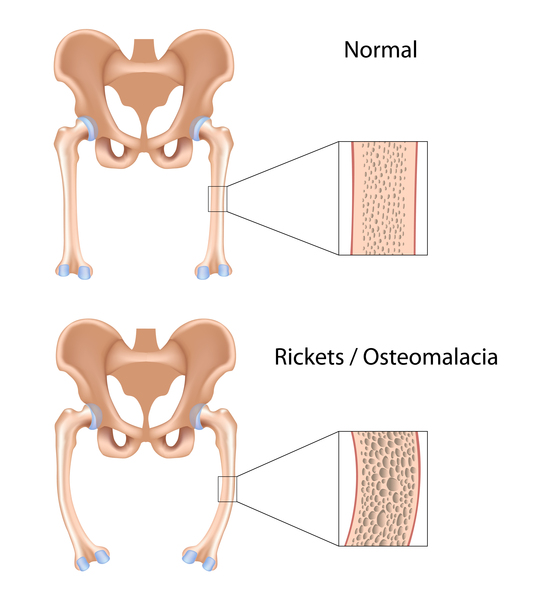
Kidney (renal) abnormalities, most commonly a condition known as renal Fanconi syndrome, frequently develop in individuals with Lowe syndrome. The kidneys play an essential role in maintaining the right amounts of minerals, salts, water, and other substances in the body. In individuals with renal Fanconi syndrome, the kidneys are unable to reabsorb important nutrients into the bloodstream. Instead, the nutrients are excreted in the urine. These kidney problems lead to increased urination, dehydration, and abnormally acidic blood (metabolic acidosis). A loss of salts and nutrients may also impair growth and result in soft, bowed bones (hypophosphatemic rickets), especially in the legs. Progressive kidney problems in older children and adults with Lowe syndrome can lead to life-threatening renal failure and end-stage renal disease (ESRD).
Frequency
Lowe syndrome is an uncommon condition. It has an estimated prevalence of 1 in 500,000 people.
Causes
Mutations in the OCRL gene cause Lowe syndrome. The OCRL gene provides instructions for making an enzyme that helps modify fat (lipid) molecules called membrane phospholipids. By controlling the levels of specific membrane phospholipids, the OCRL enzyme helps regulate the transport of certain substances to and from the cell membrane. This enzyme is also involved in the regulation of the actin cytoskeleton, which is a network of fibers that make up the cell's structural framework. The actin cytoskeleton has several critical functions, including determining cell shape and allowing cells to move.

Some mutations in the OCRL gene prevent the production of any OCRL enzyme. Other mutations reduce or eliminate the activity of the enzyme or prevent it from interacting with other proteins within the cell. Researchers are working to determine how OCRL mutations cause the characteristic features of Lowe syndrome. Because the OCRL enzyme is present throughout the body, it is unclear why the medical problems associated with this condition are mostly limited to the brain, kidneys, and eyes. It is possible that other enzymes may be able to compensate for the defective OCRL enzyme in unaffected tissues.
Inheritance
This condition is inherited in an X-linked pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation must be present in both copies of the gene to cause the disorder. Most X-linked disorders affect males much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In some cases of Lowe syndrome, an affected male inherits the mutation from a mother who carries one altered copy of the OCRL gene. Other cases result from new mutations in the gene and occur in males with no history of the disorder in their family.
Females who carry one mutated copy of the OCRL gene do not have the characteristic features of Lowe syndrome. Most female carriers, however, have changes in the lens of the eye that can be observed with a thorough eye examination. These changes typically do not impair vision.
Other Names for This Condition
- Cerebrooculorenal syndrome
- Lowe oculocerebrorenal syndrome
- Oculocerebrorenal syndrome
- Oculocerebrorenal syndrome of Lowe
- Phosphatidylinositol-4,5-bisphosphate-5-phosphatase deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, McInnes RR, Nussbaum RL. The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 1992 Jul 16;358(6383):239-42. doi: 10.1038/358239a0. Citation on PubMed
- Erdmann KS, Mao Y, McCrea HJ, Zoncu R, Lee S, Paradise S, Modregger J, Biemesderfer D, Toomre D, De Camilli P. A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell. 2007 Sep;13(3):377-90. doi: 10.1016/j.devcel.2007.08.004. Citation on PubMed or Free article on PubMed Central
- Lewis RA, Nussbaum RL, Brewer ED. Lowe Syndrome. 2001 Jul 24 [updated 2019 Apr 18]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1480/ Citation on PubMed
- Loi M. Lowe syndrome. Orphanet J Rare Dis. 2006 May 18;1:16. doi: 10.1186/1750-1172-1-16. Citation on PubMed or Free article on PubMed Central
- Lowe M. Structure and function of the Lowe syndrome protein OCRL1. Traffic. 2005 Sep;6(9):711-9. doi: 10.1111/j.1600-0854.2005.00311.x. Citation on PubMed
- Schurman SJ, Scheinman SJ. Inherited cerebrorenal syndromes. Nat Rev Nephrol. 2009 Sep;5(9):529-38. doi: 10.1038/nrneph.2009.124. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.