

Description
Langerhans cell histiocytosis is a disorder in which excess immune system cells called Langerhans cells build up in the body. Langerhans cells, which help regulate the immune system, are normally found throughout the body, especially in the skin, lymph nodes, spleen, lungs, liver, and bone marrow. In Langerhans cell histiocytosis, excess immature Langerhans cells usually form tumors called granulomas. Many researchers now consider Langerhans cell histiocytosis to be a form of cancer, but this classification remains controversial.
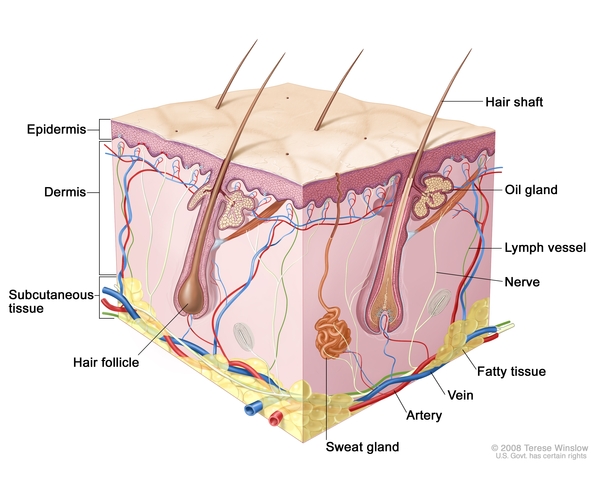
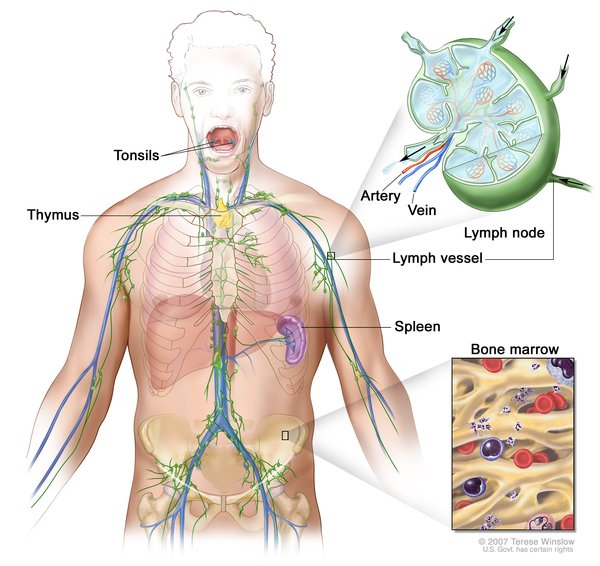

In approximately 80 percent of affected individuals, one or more granulomas develop in the bones, causing pain and swelling. The granulomas, which usually occur in the skull or the long bones of the arms or legs, may cause the bone to fracture.
Granulomas also frequently occur in the skin, appearing as blisters, reddish bumps, or rashes which can be mild to severe. The pituitary gland may also be affected; this gland is located at the base of the brain and produces hormones that control many important body functions. Without hormone supplementation, affected individuals may experience delayed or absent puberty or an inability to have children (infertility). In addition, pituitary gland damage may result in the production of excessive amounts of urine (diabetes insipidus) and dysfunction of another gland called the thyroid. Thyroid dysfunction can affect the rate of chemical reactions in the body (metabolism), body temperature, skin and hair texture, and behavior.
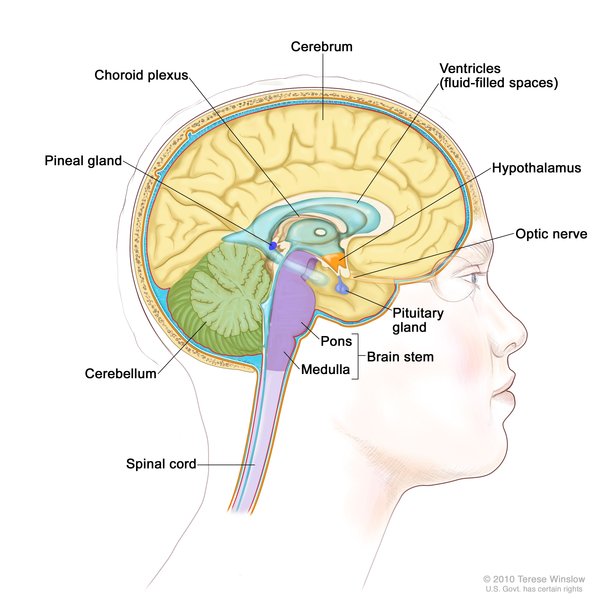
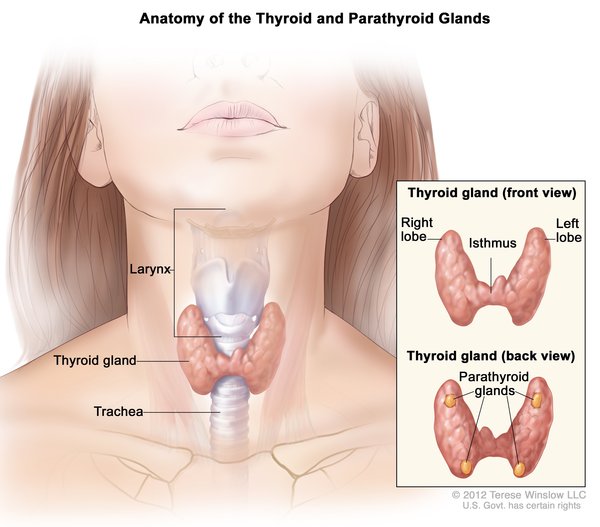
In 15 to 20 percent of cases, Langerhans cell histiocytosis affects the lungs, liver, or blood-forming (hematopoietic) system; damage to these organs and tissues may be life-threatening. Lung involvement, which appears as swelling of the small airways (bronchioles) and blood vessels of the lungs, results in stiffening of the lung tissue, breathing problems, and increased risk of infection. Hematopoietic involvement, which occurs when the Langerhans cells crowd out blood-forming cells in the bone marrow, leads to a general reduction in the number of blood cells (pancytopenia). Pancytopenia results in fatigue due to low numbers of red blood cells (anemia), frequent infections due to low numbers of white blood cells (neutropenia), and clotting problems due to low numbers of platelets (thrombocytopenia).
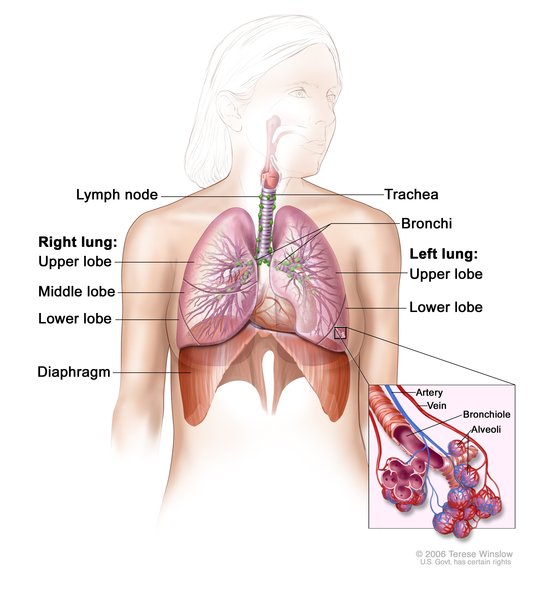


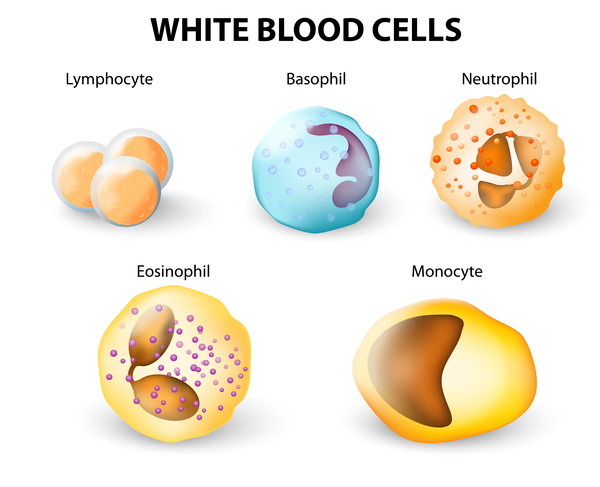
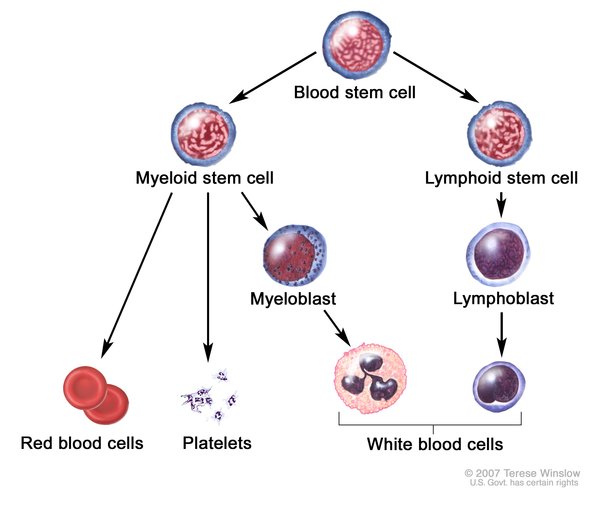
Other signs and symptoms that may occur in Langerhans cell histiocytosis, depending on which organs and tissues have Langerhans cell deposits, include swollen lymph nodes, abdominal pain, yellowing of the skin and whites of the eyes (jaundice), delayed puberty, protruding eyes, dizziness, irritability, and seizures. About 1 in 50 affected individuals experience deterioration of neurological function (neurodegeneration).
Langerhans cell histiocytosis is often diagnosed in childhood, usually between ages 2 and 3, but can appear at any age. Most individuals with adult-onset Langerhans cell histiocytosis are current or past smokers; in about two-thirds of adult-onset cases the disorder affects only the lungs.
The severity of Langerhans cell histiocytosis, and its signs and symptoms, vary widely among affected individuals. Certain presentations or forms of the disorder were formerly considered to be separate diseases. Older names that were sometimes used for forms of Langerhans cell histiocytosis include eosinophilic granuloma, Hand-Schüller-Christian disease, and Letterer-Siwe disease.
In many people with Langerhans cell histiocytosis, the disorder eventually goes away with appropriate treatment. It may even disappear on its own, especially if the disease occurs only in the skin. However, some complications of the condition, such as diabetes insipidus or other effects of tissue and organ damage, may be permanent.
Frequency
Langerhans cell histiocytosis is a rare disorder. Its prevalence is estimated at 1 to 2 in 100,000 people.
Causes
Somatic mutations in the BRAF gene have been identified in the Langerhans cells of about half of individuals with Langerhans cell histiocytosis. Somatic gene mutations are acquired during a person's lifetime and are present only in certain cells. These changes are not inherited.
The BRAF gene provides instructions for making a protein that is normally switched on and off in response to signals that control cell growth and development. Somatic mutations cause the BRAF protein in affected cells to be continuously active and to transmit messages to the nucleus even in the absence of these chemical signals. The overactive protein may contribute to the development of Langerhans cell histiocytosis by allowing the Langerhans cells to grow and divide uncontrollably.
Changes in other genes have also been identified in the Langerhans cells of some individuals with Langerhans cell histiocytosis. Some researchers believe that additional factors, such as viral infections and environmental toxins, may also influence the development of this complex disorder.
Inheritance
Langerhans cell histiocytosis is usually not inherited and typically occurs in people with no history of the disorder in their family.
A few families with multiple cases of Langerhans cell histiocytosis have been identified, but the inheritance pattern is unknown.
Other Names for This Condition
- Hashimoto-Pritzger disease
- Histiocytosis X
- Langerhans cell granulomatosis
- LCH
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Allen CE, Parsons DW. Biological and clinical significance of somatic mutations in Langerhans cell histiocytosis and related histiocytic neoplastic disorders. Hematology Am Soc Hematol Educ Program. 2015;2015:559-64. doi: 10.1182/asheducation-2015.1.559. Citation on PubMed
- Collin M, Bigley V, McClain KL, Allen CE. Cell(s) of Origin of Langerhans Cell Histiocytosis. Hematol Oncol Clin North Am. 2015 Oct;29(5):825-38. doi: 10.1016/j.hoc.2015.06.003. Epub 2015 Aug 20. Citation on PubMed or Free article on PubMed Central
- Egeler RM, Katewa S, Leenen PJ, Beverley P, Collin M, Ginhoux F, Arceci RJ, Rollins BJ; Steering Committee of the Nikolas Symposium. Langerhans cell histiocytosis is a neoplasm and consequently its recurrence is a relapse: In memory of Bob Arceci. Pediatr Blood Cancer. 2016 Oct;63(10):1704-12. doi: 10.1002/pbc.26104. Epub 2016 Jun 17. Citation on PubMed
- El Demellawy D, Young JL, de Nanassy J, Chernetsova E, Nasr A. Langerhans cell histiocytosis: a comprehensive review. Pathology. 2015 Jun;47(4):294-301. doi: 10.1097/PAT.0000000000000256. Citation on PubMed
- Grana N. Langerhans cell histiocytosis. Cancer Control. 2014 Oct;21(4):328-34. doi: 10.1177/107327481402100409. Citation on PubMed
- Harmon CM, Brown N. Langerhans Cell Histiocytosis: A Clinicopathologic Review and Molecular Pathogenetic Update. Arch Pathol Lab Med. 2015 Oct;139(10):1211-4. doi: 10.5858/arpa.2015-0199-RA. Citation on PubMed
- Monsereenusorn C, Rodriguez-Galindo C. Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis. Hematol Oncol Clin North Am. 2015 Oct;29(5):853-73. doi: 10.1016/j.hoc.2015.06.005. Epub 2015 Aug 18. Citation on PubMed
- Nelson DS, van Halteren A, Quispel WT, van den Bos C, Bovee JV, Patel B, Badalian-Very G, van Hummelen P, Ducar M, Lin L, MacConaill LE, Egeler RM, Rollins BJ. MAP2K1 and MAP3K1 mutations in Langerhans cell histiocytosis. Genes Chromosomes Cancer. 2015 Jun;54(6):361-8. doi: 10.1002/gcc.22247. Epub 2015 Mar 31. Citation on PubMed
- Roden AC, Hu X, Kip S, Parrilla Castellar ER, Rumilla KM, Vrana JA, Vassallo R, Ryu JH, Yi ES. BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol. 2014 Apr;38(4):548-51. doi: 10.1097/PAS.0000000000000129. Citation on PubMed
- Rollins BJ. Genomic Alterations in Langerhans Cell Histiocytosis. Hematol Oncol Clin North Am. 2015 Oct;29(5):839-51. doi: 10.1016/j.hoc.2015.06.004. Citation on PubMed
- Zinn DJ, Chakraborty R, Allen CE. Langerhans Cell Histiocytosis: Emerging Insights and Clinical Implications. Oncology (Williston Park). 2016 Feb;30(2):122-32, 139. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.