

Description
HSD10 disease is a disorder that affects the nervous system, vision, and heart. It is typically more severe in males than in females. Most affected males have a form of HSD10 disease in which early development seems normal, followed by a stage in which affected individuals rapidly lose skills they have acquired. This developmental regression often occurs between the ages of 1 and 2 and results in severe intellectual disability and loss of communication skills and motor skills such as sitting, standing, and walking. This form of the disorder is referred to as the infantile type. Less commonly, affected males have severe neurological problems from birth and never develop motor skills. This form is called the neonatal type. Males with the infantile or neonatal type frequently have weak muscle tone (hypotonia), recurrent seizures (epilepsy), and vision loss that gradually gets worse. Weakening of the heart muscle (cardiomyopathy) also occurs and is a common cause of death in males with severe HSD10 disease. Many affected males do not survive beyond early childhood.
Females with HSD10 disease may have developmental delay, learning problems, or intellectual disability, but they do not experience developmental regression. Some affected females have additional features of this condition, such as epilepsy, movement problems, and hearing loss. Affected females appear to have a normal life expectancy.
Frequency
HSD10 disease is a very rare disorder. Its prevalence is less than 1 in 1 million people.
Causes
HSD10 disease is caused by mutations in the HSD17B10 gene, which provides instructions for making the HSD10 protein. This protein is located within mitochondria, the energy-producing centers inside cells, where it is involved in the production (synthesis) of proteins. While most protein synthesis occurs in the fluid surrounding the nucleus (cytoplasm), a few proteins are synthesized in the mitochondria.

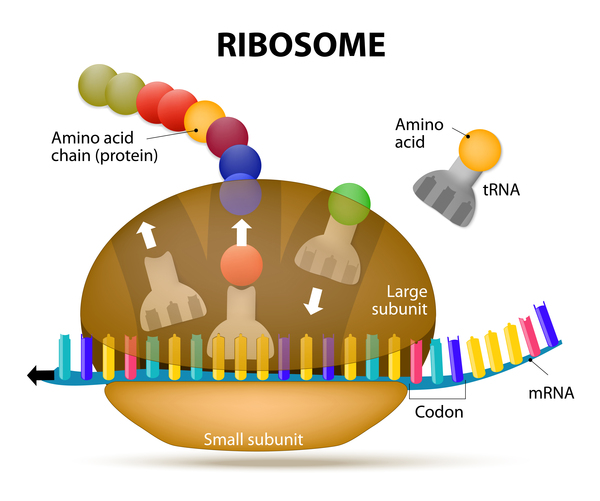
During protein synthesis, in either the mitochondria or the cytoplasm, molecules called transfer RNAs (tRNAs) help assemble protein building blocks (amino acids) into chains that form proteins. The HSD10 protein is part of a group of proteins (a complex) that is involved in making functional mitochondrial tRNA molecules, which aid in the synthesis of mitochondrial proteins. Normal mitochondrial protein production is essential for the formation of the groups of proteins that convert the energy from food into a form cells can use.

The HSD17B10 gene mutations that cause HSD10 disease reduce the amount of HSD10 protein in cells, impair their structure or function, or both, which leads to a deficiency of the functional complex in which it plays a part. This deficiency impairs the production of mitochondrial tRNAs. Without enough tRNAs, the mitochondrial synthesis of proteins involved in cellular energy production is reduced. A shortage of these proteins results in insufficient energy production in cells of the brain, eyes, and heart, leading to the characteristic features of HSD10 disease.
Inheritance
This condition is inherited in an X-linked pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males, who have only one X chromosome, a mutation in the only copy of the gene in each cell is sufficient to cause the condition. In females, who have two copies of the X chromosome, one altered copy of the gene in each cell can lead to less severe features of the condition or may cause no signs or symptoms at all. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

Other Names for This Condition
- 17β-hydroxysteroid dehydrogenase type 10 deficiency
- 2-methyl-3-hydroxybutyric aciduria
- 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency
- 2M3HBA
- 3-hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency
- 3H2MBD deficiency
- HSD10 deficiency
- Hydroxyacyl-CoA dehydrogenase II deficiency
- MHBD deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chatfield KC, Coughlin CR 2nd, Friederich MW, Gallagher RC, Hesselberth JR, Lovell MA, Ofman R, Swanson MA, Thomas JA, Wanders RJ, Wartchow EP, Van Hove JL. Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion. 2015 Mar;21:1-10. doi: 10.1016/j.mito.2014.12.005. Epub 2015 Jan 6. Citation on PubMed or Free article on PubMed Central
- Deutschmann AJ, Amberger A, Zavadil C, Steinbeisser H, Mayr JA, Feichtinger RG, Oerum S, Yue WW, Zschocke J. Mutation or knock-down of 17beta-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum Mol Genet. 2014 Jul 1;23(13):3618-28. doi: 10.1093/hmg/ddu072. Epub 2014 Feb 18. Citation on PubMed
- Holzmann J, Frank P, Loffler E, Bennett KL, Gerner C, Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008 Oct 31;135(3):462-74. doi: 10.1016/j.cell.2008.09.013. Citation on PubMed
- Ofman R, Ruiter JP, Feenstra M, Duran M, Poll-The BT, Zschocke J, Ensenauer R, Lehnert W, Sass JO, Sperl W, Wanders RJ. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency is caused by mutations in the HADH2 gene. Am J Hum Genet. 2003 May;72(5):1300-7. doi: 10.1086/375116. Epub 2003 Apr 14. Citation on PubMed or Free article on PubMed Central
- Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J, Rossmanith W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase--extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012 Dec;40(22):11583-93. doi: 10.1093/nar/gks910. Epub 2012 Oct 5. Citation on PubMed or Free article on PubMed Central
- Vilardo E, Rossmanith W. Molecular insights into HSD10 disease: impact of SDR5C1 mutations on the human mitochondrial RNase P complex. Nucleic Acids Res. 2015 Jul 27;43(13):6649. doi: 10.1093/nar/gkv658. Epub 2015 Jun 19. No abstract available. Citation on PubMed or Free article on PubMed Central
- Yang SY, He XY, Miller D. HSD17B10: a gene involved in cognitive function through metabolism of isoleucine and neuroactive steroids. Mol Genet Metab. 2007 Sep-Oct;92(1-2):36-42. doi: 10.1016/j.ymgme.2007.06.001. Epub 2007 Jul 6. Citation on PubMed
- Yang SY, He XY, Schulz H. Multiple functions of type 10 17beta-hydroxysteroid dehydrogenase. Trends Endocrinol Metab. 2005 May-Jun;16(4):167-75. doi: 10.1016/j.tem.2005.03.006. Citation on PubMed
- Zschocke J. HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J Inherit Metab Dis. 2012 Jan;35(1):81-9. doi: 10.1007/s10545-011-9415-4. Epub 2011 Nov 30. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.