

Description
Hajdu-Cheney syndrome is a rare disorder that can affect many parts of the body, particularly the bones. Bone loss from the tips of the fingers and toes (acroosteolysis) is a characteristic feature of the condition. The fingers and toes may appear short and rounded, and they may become shorter over time as the bone continues to break down. In people with Hajdu-Cheney syndrome, the fingers are more likely to be affected than the toes. Bone loss in the fingers can interfere with fine motor skills, such as picking up small objects.
The signs and symptoms of Hajdu-Cheney syndrome vary greatly among affected individuals, even among members of the same family. Many of the disorder's features, including acroosteolysis, are not present at birth but become apparent in childhood or later.
Additional bone abnormalities are common in people with Hajdu-Cheney syndrome. Affected individuals typically develop osteoporosis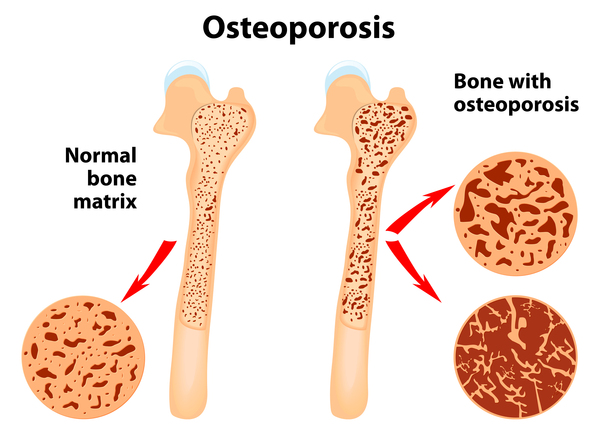 , which causes the bones to be brittle and prone to fracture. Many affected individuals experience a type of fracture called a compression fracture in the spinal bones (vertebrae). Some develop an abnormal curvature of the spine (scoliosis or kyphosis
, which causes the bones to be brittle and prone to fracture. Many affected individuals experience a type of fracture called a compression fracture in the spinal bones (vertebrae). Some develop an abnormal curvature of the spine (scoliosis or kyphosis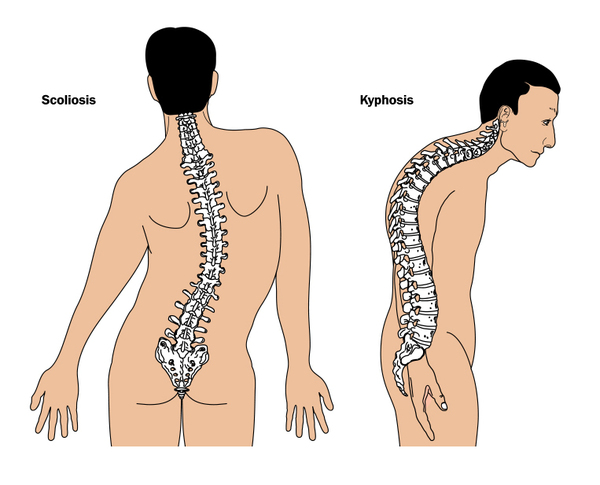 ). Hajdu-Cheney syndrome can also affect the shape and strength of the long bones in the arms and legs. The bone abnormalities that are associated with this condition often lead to short stature.
). Hajdu-Cheney syndrome can also affect the shape and strength of the long bones in the arms and legs. The bone abnormalities that are associated with this condition often lead to short stature.
Hajdu-Cheney syndrome can also involve the bones of the skull. The shape of the skull is often dolichocephalic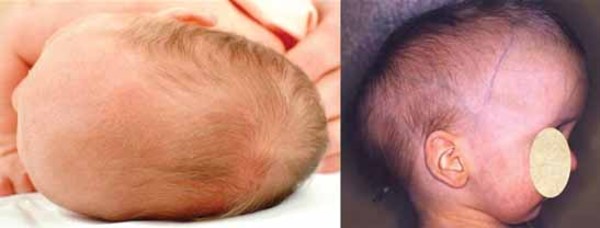 , which means it is elongated from back to front. In many affected individuals, the bone at the back of the skull bulges outward, causing a bump called a prominent occiput
, which means it is elongated from back to front. In many affected individuals, the bone at the back of the skull bulges outward, causing a bump called a prominent occiput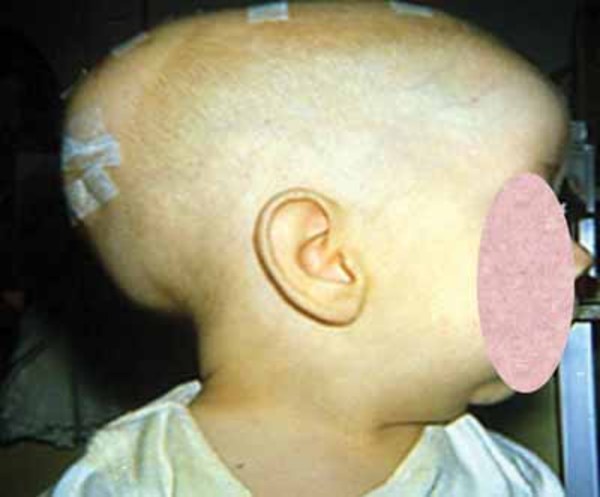 . Serious complications of Hajdu-Cheney syndrome include abnormalities known as platybasia and basilar invagination. Platybasia is a flattening of the base of the skull that is caused by thinning and softening of the skull bones. Basilar invagination occurs when the softened bones allow part of the spine to protrude through the opening at the bottom of the skull and push into the lower parts of the brain. These abnormalities can lead to severe neurological problems, such as a buildup of fluid in the brain (hydrocephalus) or spinal cord (syringomyelia) and breathing difficulties. In some cases, these abnormalities can be life-threatening.
. Serious complications of Hajdu-Cheney syndrome include abnormalities known as platybasia and basilar invagination. Platybasia is a flattening of the base of the skull that is caused by thinning and softening of the skull bones. Basilar invagination occurs when the softened bones allow part of the spine to protrude through the opening at the bottom of the skull and push into the lower parts of the brain. These abnormalities can lead to severe neurological problems, such as a buildup of fluid in the brain (hydrocephalus) or spinal cord (syringomyelia) and breathing difficulties. In some cases, these abnormalities can be life-threatening.
Additional signs and symptoms can include a small lower jaw (micrognathia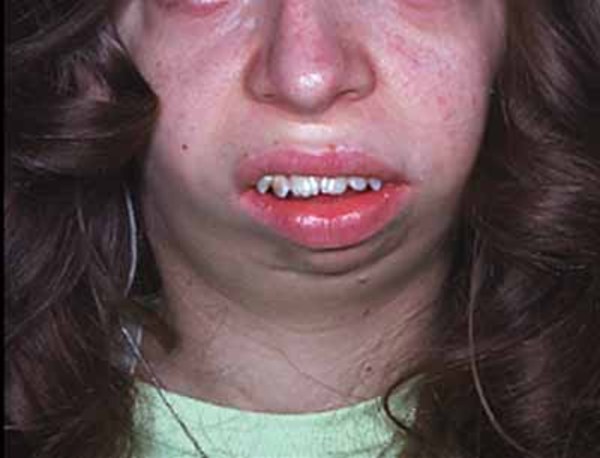 ) and an opening in the roof of the mouth called a cleft palate
) and an opening in the roof of the mouth called a cleft palate . Adults with Hajdu-Cheney syndrome may have facial features that are described as "coarse." These facial features can include widely spaced
. Adults with Hajdu-Cheney syndrome may have facial features that are described as "coarse." These facial features can include widely spaced and downward-slanting
and downward-slanting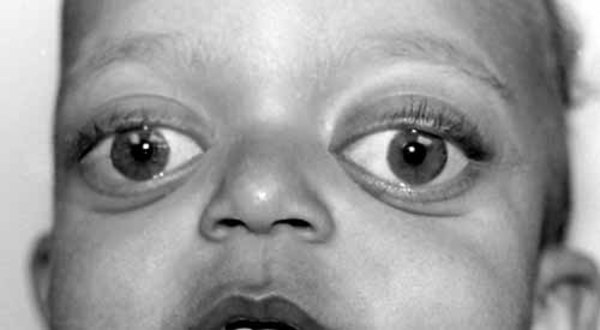 eyes, eyebrows that grow together in the middle (synophrys
eyes, eyebrows that grow together in the middle (synophrys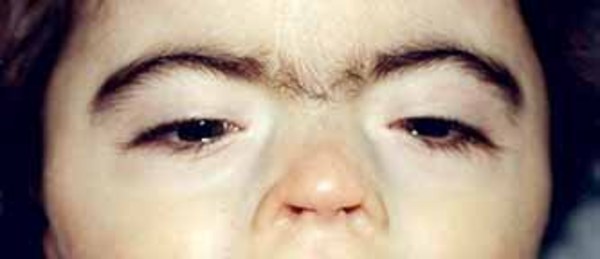 ), low-set ears, and a large space between the nose and upper lip (a long philtrum
), low-set ears, and a large space between the nose and upper lip (a long philtrum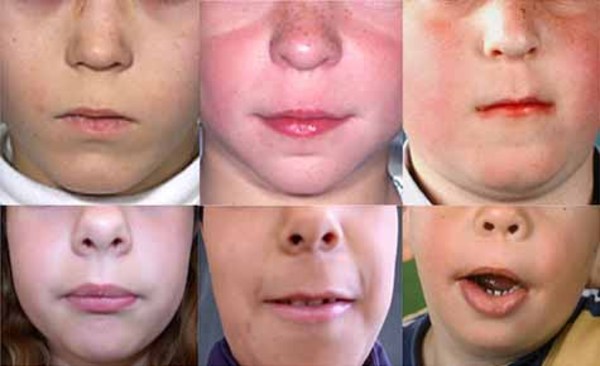 ).
).
Other features of Hajdu-Cheney syndrome can include joint abnormalities, particularly an unusually large range of joint movement (hypermobility); dental problems; hearing loss; and a deep, gravelly voice. Affected individuals may also have recurrent childhood infections, heart defects, and kidney abnormalities that include the growth of multiple fluid-filled cysts (polycystic kidneys ). Some people with this condition have delayed development in childhood, but the delays are usually mild.
). Some people with this condition have delayed development in childhood, but the delays are usually mild.
Frequency
Although Hajdu-Cheney syndrome is a rare disease, its exact prevalence is unknown. Fewer than 100 affected individuals have been described in the medical literature.
Causes
Hajdu-Cheney syndrome is associated with variants (also called mutations) in the NOTCH2 gene. This gene provides instructions for making a receptor protein. Other proteins, called ligands, can fit into specific sites on receptor proteins, like a key into a lock. When a ligand binds to the NOTCH2 receptor protein, it triggers a signaling pathway called the NOTCH2 signaling pathway. NOTCH2 signaling is important for the early development of bones and for bone remodeling, a normal process in which old bone is removed and new bone is created to replace it. NOTCH2 signaling also appears to be involved in the development of the heart, kidneys, teeth, and other parts of the body.
Variants in a specific area near the end of the NOTCH2 gene are associated with Hajdu-Cheney syndrome. These variants cause cells to produce a version of the NOTCH2 receptor protein that cannot be broken down normally. As a result, the protein accumulates, which leads to an increase in NOTCH2 signaling. It is unclear exactly how the buildup of NOTCH2 receptor proteins leads to the various features of Hajdu-Cheney syndrome. Researchers suspect that the skeletal features of the disorder, including acroosteolysis, osteoporosis, and the appearance of certain facial features, are a result of abnormal bone development and remodeling.
Inheritance
Hajdu-Cheney syndrome is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder. Many cases of this condition result from a new (de novo) variant in the gene that occurs during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or during early embryonic development. These affected individuals typically have no history of the disorder in their family.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Many cases of this condition result from a new (de novo) variant in the gene that occurs during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or during early embryonic development. These affected individuals typically have no history of the disorder in their family.
Other Names for This Condition
- Acroosteolysis dominant type
- Acroosteolysis with osteoporosis and changes in skull and mandible
- Arthrodentoosteodysplasia
- Cheney syndrome
- Hajdu-cheney syndrome-notch2
- HJCYS
- Serpentine fibula polycystic kidney syndrome
- SFPKS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Abdelkarim M, Alageel D, Ahsan F, Alhuthil R, Alsarhani H, Alsagheir A. Hajdu-Cheney syndrome with a novel variant in NOTCH2 gene: A case report. Bone Rep. 2023 Aug 18;19:101709. doi: 10.1016/j.bonr.2023.101709. eCollection 2023 Dec. Citation on PubMed
- Brennan AM, Pauli RM. Hajdu--Cheney syndrome: evolution of phenotype and clinical problems. Am J Med Genet. 2001 May 15;100(4):292-310. doi: 10.1002/1096-8628(20010515)100:43.0.co;2-4. Citation on PubMed
- Canalis E, Zanotti S. Hajdu-Cheney syndrome: a review. Orphanet J Rare Dis. 2014 Dec 10;9:200. doi: 10.1186/s13023-014-0200-y. Citation on PubMed or Free article on PubMed Central
- Canalis E. Clinical and experimental aspects of notch receptor signaling: Hajdu-Cheney syndrome and related disorders. Metabolism. 2018 Mar;80:48-56. doi: 10.1016/j.metabol.2017.08.002. Epub 2017 Aug 24. Citation on PubMed
- Cortes-Martin J, Diaz-Rodriguez L, Piqueras-Sola B, Rodriguez-Blanque R, Bermejo-Fernandez A, Sanchez-Garcia JC. Hajdu-Cheney Syndrome: A Systematic Review of the Literature. Int J Environ Res Public Health. 2020 Aug 25;17(17):6174. doi: 10.3390/ijerph17176174. Citation on PubMed
- Gray MJ, Kim CA, Bertola DR, Arantes PR, Stewart H, Simpson MA, Irving MD, Robertson SP. Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome. Eur J Hum Genet. 2012 Jan;20(1):122-4. doi: 10.1038/ejhg.2011.125. Epub 2011 Jun 29. Citation on PubMed or Free article on PubMed Central
- Isidor B, Lindenbaum P, Pichon O, Bezieau S, Dina C, Jacquemont S, Martin-Coignard D, Thauvin-Robinet C, Le Merrer M, Mandel JL, David A, Faivre L, Cormier-Daire V, Redon R, Le Caignec C. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat Genet. 2011 Mar 6;43(4):306-8. doi: 10.1038/ng.778. Citation on PubMed
- Majewski J, Schwartzentruber JA, Caqueret A, Patry L, Marcadier J, Fryns JP, Boycott KM, Ste-Marie LG, McKiernan FE, Marik I, Van Esch H; FORGE Canada Consortium; Michaud JL, Samuels ME. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum Mutat. 2011 Oct;32(10):1114-7. doi: 10.1002/humu.21546. Epub 2011 Sep 9. Citation on PubMed
- Narumi Y, Min BJ, Shimizu K, Kazukawa I, Sameshima K, Nakamura K, Kosho T, Rhee Y, Chung YS, Kim OH, Fukushima Y, Park WY, Nishimura G. Clinical consequences in truncating mutations in exon 34 of NOTCH2: report of six patients with Hajdu-Cheney syndrome and a patient with serpentine fibula polycystic kidney syndrome. Am J Med Genet A. 2013 Mar;161A(3):518-26. doi: 10.1002/ajmg.a.35772. Epub 2013 Feb 7. Citation on PubMed
- Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Mansour S, Holder SE, Brain CE, Burton BK, Kim KH, Pauli RM, Aftimos S, Stewart H, Kim CA, Holder-Espinasse M, Robertson SP, Drake WM, Trembath RC. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet. 2011 Mar 6;43(4):303-5. doi: 10.1038/ng.779. Citation on PubMed
- Zhao W, Petit E, Gafni RI, Collins MT, Robey PG, Seton M, Miller KK, Mannstadt M. Mutations in NOTCH2 in patients with Hajdu-Cheney syndrome. Osteoporos Int. 2013 Aug;24(8):2275-81. doi: 10.1007/s00198-013-2298-5. Epub 2013 Feb 7. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.