

Description
GRACILE syndrome is a severe disorder that begins before birth. GRACILE stands for the condition's characteristic features: growth retardation, aminoaciduria, cholestasis, iron overload, lactic acidosis, and early death.
In GRACILE syndrome, growth before birth is slow (intrauterine growth retardation). Affected newborns are smaller than average and have an inability to grow and gain weight at the expected rate (failure to thrive). A characteristic of GRACILE syndrome is excess iron in the liver, which likely begins before birth. Iron levels may begin to improve after birth, although they typically remain elevated. Within the first day of life, infants with GRACILE syndrome have a buildup of a chemical called lactic acid in the body (lactic acidosis). They also have kidney problems that lead to an excess of molecules called amino acids in the urine (aminoaciduria). Babies with GRACILE syndrome have cholestasis, which is a reduced ability to produce and release a digestive fluid called bile. Cholestasis leads to irreversible liver disease (cirrhosis) in the first few months of life.
Because of the severe health problems caused by GRACILE syndrome, infants with this condition do not survive for more than a few months, and about half die within a few days of birth.
Frequency
GRACILE syndrome is found almost exclusively in Finland, where it is estimated to affect 1 in 47,000 infants. At least 32 affected infants have been described in the medical literature.
Causes
GRACILE syndrome is caused by a mutation in the BCS1L gene. The protein produced from this gene is found in cell structures called mitochondria, which convert the energy from food into a form that cells can use. In mitochondria, the BCS1L protein plays a role in oxidative phosphorylation, which is a multistep process through which cells derive much of their energy. The BCS1L protein is critical for the formation of a group of proteins known as complex III, which is one of several protein complexes involved in oxidative phosphorylation.

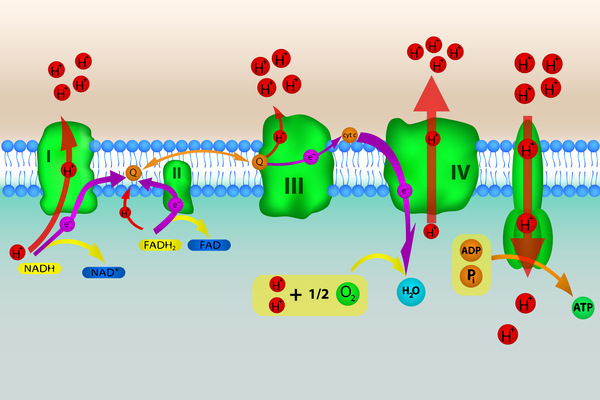
The genetic change involved in GRACILE syndrome alters the BCS1L protein, and the abnormal protein is broken down more quickly than the normal protein. What little protein remains is able to help form some complete complex III, although the amount is severely reduced, particularly in the liver and kidneys. As a result, complex III activity and oxidative phosphorylation are decreased in these organs in people with GRACILE syndrome. Without energy, these organs become damaged, leading to many of the features of GRACILE syndrome. It is not clear why a change in the BCS1L gene leads to iron accumulation in people with this condition.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Fellman syndrome
- Finnish lactic acidosis with hepatic hemosiderosis
- Finnish lethal neonatal metabolic syndrome
- Growth retardation, amino aciduria, cholestasis, iron overload, lactic acidosis, and early death
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Fellman V. The GRACILE syndrome, a neonatal lethal metabolic disorder with iron overload. Blood Cells Mol Dis. 2002 Nov-Dec;29(3):444-50. doi: 10.1006/bcmd.2002.0582. Citation on PubMed
- Kotarsky H, Karikoski R, Morgelin M, Marjavaara S, Bergman P, Zhang DL, Smet J, van Coster R, Fellman V. Characterization of complex III deficiency and liver dysfunction in GRACILE syndrome caused by a BCS1L mutation. Mitochondrion. 2010 Aug;10(5):497-509. doi: 10.1016/j.mito.2010.05.009. Epub 2010 May 23. Citation on PubMed
- Visapaa I, Fellman V, Vesa J, Dasvarma A, Hutton JL, Kumar V, Payne GS, Makarow M, Van Coster R, Taylor RW, Turnbull DM, Suomalainen A, Peltonen L. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet. 2002 Oct;71(4):863-76. doi: 10.1086/342773. Epub 2002 Sep 5. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.