

Description
Glycogen storage disease type VII (GSDVII) is an inherited disorder caused by an inability to break down a complex sugar called glycogen in muscle cells. A lack of glycogen breakdown interferes with the function of muscle cells.
There are four types of GSDVII. They are differentiated by their signs and symptoms and the age at which symptoms first appear.
The classical form of GSDVII is the most common form. Its features usually appear in childhood. This form is characterized by muscle pain and cramps, often following moderate exercise; strenuous exercise can lead to nausea and vomiting. During exercise, muscle tissue can be abnormally broken down, releasing a protein called myoglobin. This protein is processed by the kidneys and released in the urine (myoglobinuria). If untreated, myoglobinuria can damage the kidneys and lead to kidney failure. Some people with the classical form of GSDVII develop high levels of a waste product called uric acid in the blood (hyperuricemia) because the damaged kidneys are unable to remove uric acid effectively. Affected individuals may also have elevated levels of a molecule called bilirubin in the blood that can cause yellowing of the skin and whites of the eyes (jaundice). Individuals with classical GSDVII often have elevated levels of an enzyme called creatine kinase in their blood. This finding is a common indicator of muscle disease.

Infants with the severe infantile form of GSDVII have low muscle tone (hypotonia) at birth, which leads to muscle weakness (myopathy) that worsens over time. Affected infants have a weakened and enlarged heart (cardiomyopathy) and difficulty breathing normally. Individuals with this form of GSDVII usually do not survive past their first year of life.
In the late-onset form of GSDVII, myopathy is typically the only feature. The muscle weakness appears in adulthood, although some individuals have difficulty with sustained exercise starting in childhood. The weakness generally affects the muscles closest to the center of the body (proximal muscles).
The hemolytic form of GSDVII is characterized by hemolytic anemia, in which red blood cells are broken down (undergo hemolysis) prematurely, causing a shortage of red blood cells (anemia). People with the hemolytic form of GSDVII do not experience any signs or symptoms of muscle pain or weakness related to the disorder.

Frequency
GSDVII is thought to be a rare condition; more than 100 cases have been described in the scientific literature.
Causes
Mutations in the PFKM gene cause GSDVII. This gene provides instructions for making one piece (the PFKM subunit) of an enzyme called phosphofructokinase, which plays a role in the breakdown of glycogen. The phosphofructokinase enzyme is made up of four subunits and is found in a variety of tissues. Different combinations of subunits are found in different tissues. In muscles used for movement (skeletal muscles), the phosphofructokinase enzyme is composed solely of PFKM subunits.
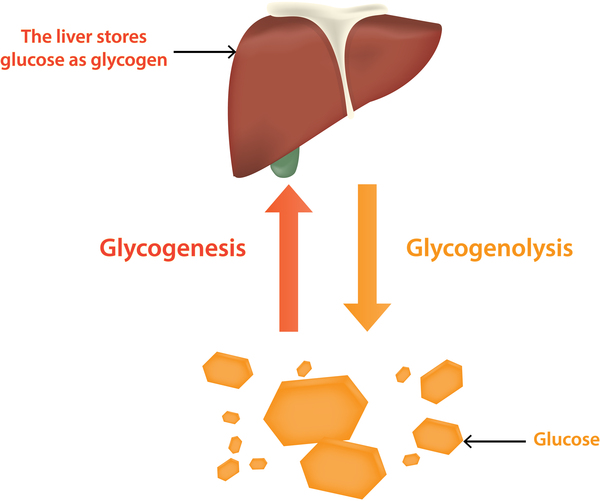
In skeletal muscle, the cells' main source of energy is stored as glycogen. Glycogen can be broken down rapidly into the simple sugar glucose when energy is needed, for instance to maintain normal blood glucose levels between meals or for energy during exercise. Phosphofructokinase is involved in the sequence of events that breaks down glycogen to provide energy to muscle cells.
PFKM gene mutations result in the production of PFKM subunits that have little or no function. As a result, no functional phosphofructokinase is formed in skeletal muscles, and glycogen cannot be completely broken down. Partially broken down glycogen then builds up in muscle cells. Muscles that do not have access to glycogen as an energy source become weakened and cramped following moderate strain, such as exercise, and in some cases, begin to break down. In other tissues, other subunits that make up the phosphofructokinase enzyme likely compensate for the lack of PFKM subunits, and the enzyme is able to retain some function. This compensation may help explain why other tissues are not affected by PFKM gene mutations. It is unclear why some individuals with GSDVII are affected with more severe forms of the disorder than others.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Glycogenosis 7
- GSD VII
- GSD7
- Muscle phosphofructokinase deficiency
- PFKM deficiency
- Phosphofructokinase deficiency
- Tarui disease
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bruser A, Kirchberger J, Schoneberg T. Altered allosteric regulation of muscle 6-phosphofructokinase causes Tarui disease. Biochem Biophys Res Commun. 2012 Oct 12;427(1):133-7. doi: 10.1016/j.bbrc.2012.09.024. Epub 2012 Sep 17. Citation on PubMed
- Di Mauro S. Muscle glycogenoses: an overview. Acta Myol. 2007 Jul;26(1):35-41. No abstract available. Citation on PubMed or Free article on PubMed Central
- Musumeci O, Bruno C, Mongini T, Rodolico C, Aguennouz M, Barca E, Amati A, Cassandrini D, Serlenga L, Vita G, Toscano A. Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscul Disord. 2012 Apr;22(4):325-30. doi: 10.1016/j.nmd.2011.10.022. Epub 2011 Nov 30. Citation on PubMed
- Toscano A, Musumeci O. Tarui disease and distal glycogenoses: clinical and genetic update. Acta Myol. 2007 Oct;26(2):105-7. Citation on PubMed or Free article on PubMed Central
- Vives-Corrons JL, Koralkova P, Grau JM, Manu Pereira Mdel M, Van Wijk R. First description of phosphofructokinase deficiency in spain: identification of a novel homozygous missense mutation in the PFKM gene. Front Physiol. 2013 Dec 30;4:393. doi: 10.3389/fphys.2013.00393. eCollection 2013. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.