

Description
Glycogen storage disease type I (also known as GSDI or von Gierke disease) is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body's cells. The accumulation of glycogen in certain organs and tissues, especially the liver, kidneys, and small intestines, impairs their ability to function normally.
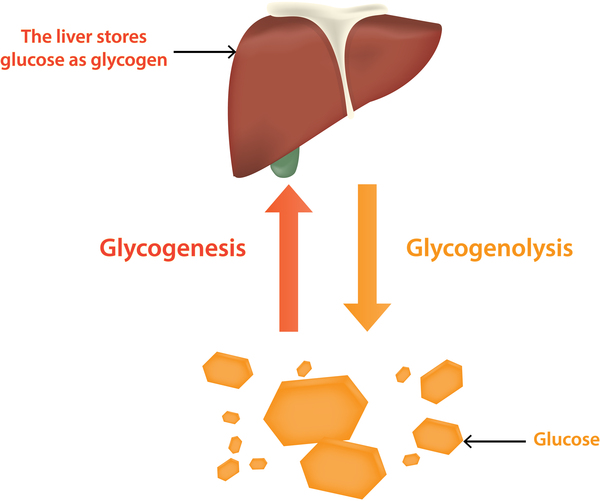
Signs and symptoms of this condition typically appear around the age of 3 or 4 months, when babies start to sleep through the night and do not eat as frequently as newborns. Affected infants may have low blood sugar (hypoglycemia), which can lead to seizures. They can also have a buildup of lactic acid in the body (lactic acidosis), high blood levels of a waste product called uric acid (hyperuricemia), and excess amounts of fats in the blood (hyperlipidemia). As they get older, children with GSDI have thin arms and legs and short stature. An enlarged liver may give the appearance of a protruding abdomen. The kidneys may also be enlarged. Affected individuals may also have diarrhea and deposits of cholesterol in the skin (xanthomas).
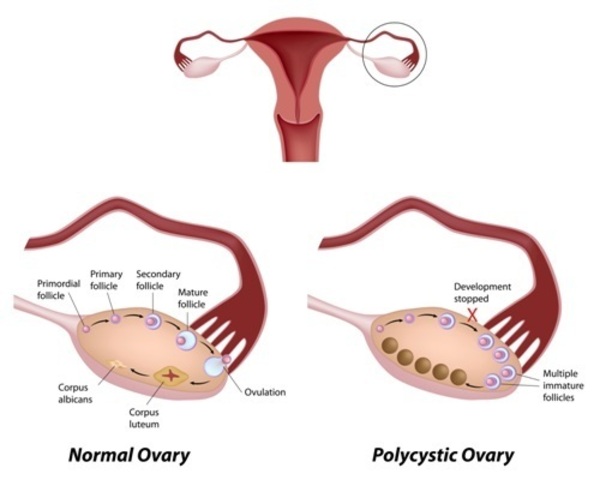
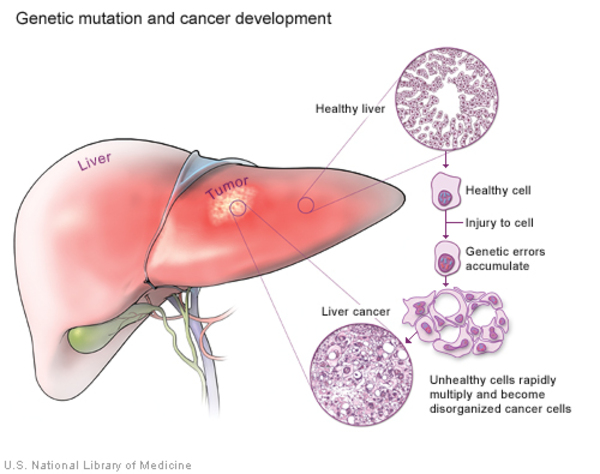
People with GSDI may experience delayed puberty. Beginning in young to mid-adulthood, affected individuals may have thinning of the bones (osteoporosis), a form of arthritis resulting from uric acid crystals in the joints (gout), kidney disease, and high blood pressure in the blood vessels that supply the lungs (pulmonary hypertension). Females with this condition may also have abnormal development of the ovaries (polycystic ovaries). In affected teens and adults, tumors called adenomas may form in the liver. Adenomas are usually noncancerous (benign), but occasionally these tumors can become cancerous (malignant).
Researchers have described two types of GSDI, which differ in their signs and symptoms and genetic cause. These types are known as glycogen storage disease type Ia (GSDIa) and glycogen storage disease type Ib (GSDIb). Two other forms of GSDI have been described, and they were originally named types Ic and Id. However, these types are now known to be variations of GSDIb; for this reason, GSDIb is sometimes called GSD type I non-a.
Many people with GSDIb have a shortage of white blood cells (neutropenia), which can make them prone to recurrent bacterial infections. Neutropenia is usually apparent by age 1. Many affected individuals also have inflammation of the intestinal walls (inflammatory bowel disease). People with GSDIb may have oral problems including cavities, inflammation of the gums (gingivitis), chronic gum (periodontal) disease, abnormal tooth development, and open sores (ulcers) in the mouth. The neutropenia and oral problems are specific to people with GSDIb and are typically not seen in people with GSDIa.
Frequency
The overall incidence of GSDI is 1 in 100,000 individuals. GSDIa is more common than GSDIb, accounting for 80 percent of all GSDI cases.
Causes
Variants (also called mutations) in two genes, G6PC1 and SLC37A4, cause GSDI. G6PC1 gene variants cause GSDIa, and SLC37A4 gene variants cause GSDIb.
The proteins produced from the G6PC1 and SLC37A4 genes work together to break down a type of sugar molecule called glucose 6-phosphate. The breakdown of this molecule produces the simple sugar glucose, which is the primary energy source for most cells in the body.
Variants in the G6PC1 and SLC37A4 genes prevent the effective breakdown of glucose 6-phosphate. Glucose 6-phosphate that is not broken down to glucose is converted to glycogen and fat so it can be stored within cells. Too much glycogen and fat stored within a cell can be toxic. This buildup damages organs and tissues throughout the body, particularly the liver and kidneys, leading to the signs and symptoms of GSDI.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Glucose-6-phosphate deficiency
- Glucose-6-phosphate transport defect
- GSD I
- GSD type I
- Hepatorenal form of glycogen storage disease
- Hepatorenal glycogenosis
- Von Gierke disease
- Von Gierke's disease
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bali DS, El-Gharbawy A, Austin S, Pendyal S, Kishnani PS. Glycogen Storage Disease Type I. 2006 Apr 19 [updated 2021 Oct 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1312/ Citation on PubMed
- Chou JY, Jun HS, Mansfield BC. Neutropenia in type Ib glycogen storage disease. Curr Opin Hematol. 2010 Jan;17(1):36-42. doi: 10.1097/MOH.0b013e328331df85. Citation on PubMed or Free article on PubMed Central
- Chou JY, Mansfield BC. Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease. Hum Mutat. 2008 Jul;29(7):921-30. doi: 10.1002/humu.20772. Citation on PubMed or Free article on PubMed Central
- Froissart R, Piraud M, Boudjemline AM, Vianey-Saban C, Petit F, Hubert-Buron A, Eberschweiler PT, Gajdos V, Labrune P. Glucose-6-phosphatase deficiency. Orphanet J Rare Dis. 2011 May 20;6:27. doi: 10.1186/1750-1172-6-27. Citation on PubMed or Free article on PubMed Central
- Kishnani PS, Austin SL, Abdenur JE, Arn P, Bali DS, Boney A, Chung WK, Dagli AI, Dale D, Koeberl D, Somers MJ, Wechsler SB, Weinstein DA, Wolfsdorf JI, Watson MS; American College of Medical Genetics and Genomics. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 2014 Nov;16(11):e1. doi: 10.1038/gim.2014.128. Citation on PubMed
- Melis D, Fulceri R, Parenti G, Marcolongo P, Gatti R, Parini R, Riva E, Della Casa R, Zammarchi E, Andria G, Benedetti A. Genotype/phenotype correlation in glycogen storage disease type 1b: a multicentre study and review of the literature. Eur J Pediatr. 2005 Aug;164(8):501-8. doi: 10.1007/s00431-005-1657-4. Epub 2005 May 19. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.