

Description
Fryns syndrome is a condition that affects the development of many parts of the body.
Most people with Fryns syndrome have a defect in the muscle that separates the abdomen from the chest cavity (the diaphragm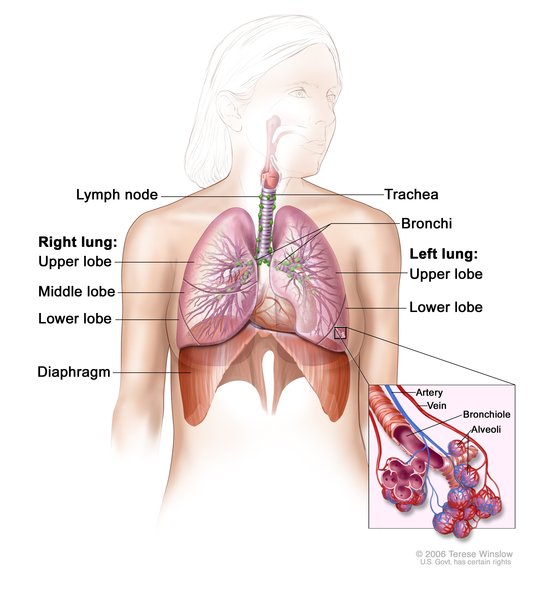 ). The most common defect is a congenital diaphragmatic hernia, which is a hole in the diaphragm that develops before birth. This hole allows the stomach and intestines to move into the chest and crowd the heart and lungs. As a result, the lungs often do not develop properly (pulmonary hypoplasia), which can cause life-threatening breathing difficulties in affected infants.
). The most common defect is a congenital diaphragmatic hernia, which is a hole in the diaphragm that develops before birth. This hole allows the stomach and intestines to move into the chest and crowd the heart and lungs. As a result, the lungs often do not develop properly (pulmonary hypoplasia), which can cause life-threatening breathing difficulties in affected infants.
People with Fryns syndrome typically have abnormalities of the fingers and toes and distinctive facial features. The tips of the fingers and toes
and toes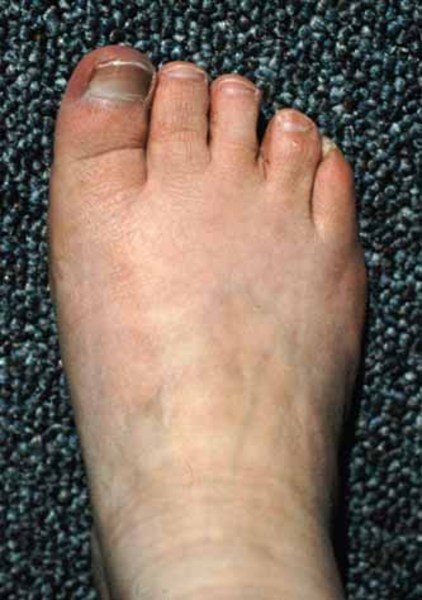 tend to be underdeveloped, so they can look short and stubby and have small nails or no nails at all. Affected individuals may have widely spaced eyes (hypertelorism
tend to be underdeveloped, so they can look short and stubby and have small nails or no nails at all. Affected individuals may have widely spaced eyes (hypertelorism ), a broad and flat nasal bridge
), a broad and flat nasal bridge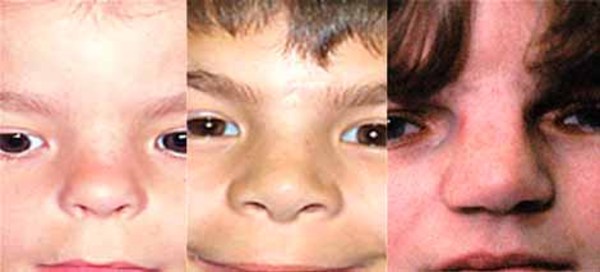 , a thick nasal tip, a long space between the nose and upper lip (a long philtrum
, a thick nasal tip, a long space between the nose and upper lip (a long philtrum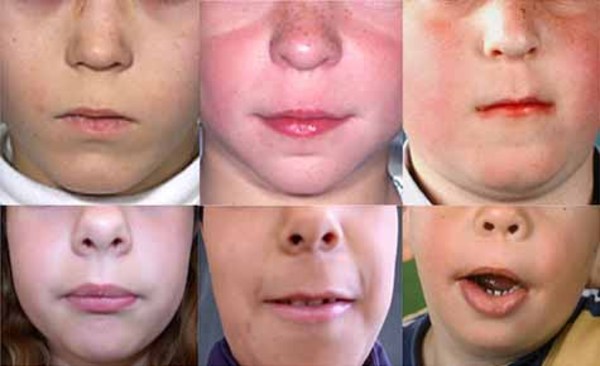 ), a large mouth (macrostomia
), a large mouth (macrostomia ), and a small lower jaw (micrognathia
), and a small lower jaw (micrognathia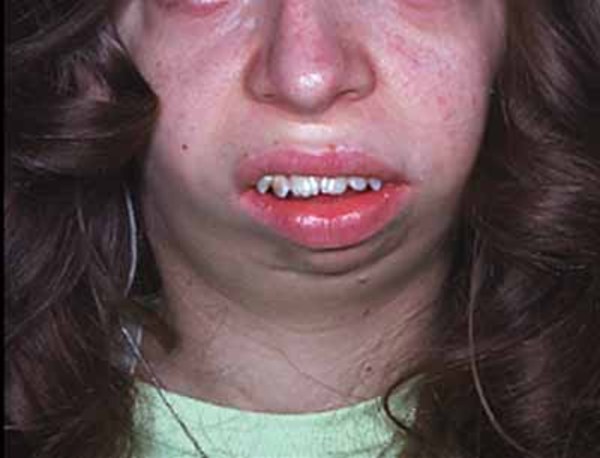 ). Many also have low-set and abnormally shaped ears.
). Many also have low-set and abnormally shaped ears.
Additional features of Fryns syndrome include small eyes (microphthalmia), clouding of the clear outer covering of the eye (the cornea), and an opening in the roof of the mouth (cleft palate
(the cornea), and an opening in the roof of the mouth (cleft palate ) with or without a split in the lip (cleft lip
) with or without a split in the lip (cleft lip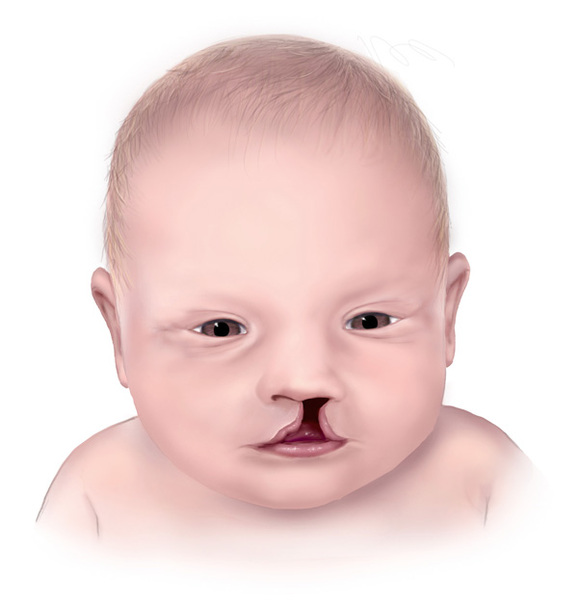 ). Fryns syndrome can also affect the development of the brain
). Fryns syndrome can also affect the development of the brain , heart, digestive system, kidneys
, heart, digestive system, kidneys , and genitalia.
, and genitalia.
Because there are significant health problems associated with congenital diaphragmatic hernias, most children with Fryns syndrome do not survive past infancy. Affected individuals who do survive into childhood often have severe developmental delays and intellectual disabilities.
The features of this condition often overlap with those of other disorders. As a result, Fryns syndrome can be difficult to diagnose.
Frequency
The exact prevalence of Fryns syndrome is unknown; fewer than 100 individuals with this condition have been reported in the medical literature. Studies suggest that up to 10 percent of all cases of congenital diaphragmatic hernia may be due to Fryns syndrome.
Causes
Variants (also called mutations) in the PIGN gene have been found to cause some cases of Fryns syndrome. The PIGN gene provides instructions for making an enzyme called GPI ethanolamine phosphate transferase 1. This enzyme takes part in a series of steps that produce a molecule called a glycophosphatidylinositol (GPI) anchor. The GPI anchor transports many different proteins to the cell membrane, ensuring that these proteins are available when needed.
The PIGN gene variants that cause Fryns syndrome are known as "loss-of-function variants" because they reduce the amount of functional GPI ethanolamine phosphate transferase 1 enzyme that is available to modify the GPI anchor. As a result, the GPI anchor cannot deliver proteins to their proper places on the cell membrane. This disrupts critical developmental pathways, which leads to the signs and symptoms seen in people with Fryns syndrome.
Researchers are trying to determine other possible genetic causes for Fryns syndrome. Chromosomal changes, such as missing (deleted) chromosomal material, that can cause features similar to those seen in people with Fryns syndrome are of particular interest to investigators.
Inheritance
Fryns syndrome appears to be inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Diaphragmatic hernia, abnormal face, and distal limb anomalies
- Diaphragmatic hernia-facial dysmorphism-distal limb anomalies syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Alessandri JL, Gordon CT, Jacquemont ML, Gruchy N, Ajeawung NF, Benoist G, Oufadem M, Chebil A, Duffourd Y, Dumont C, Gerard M, Kuentz P, Jouan T, Filippini F, Nguyen TTM, Alibeu O, Bole-Feysot C, Nitschke P, Omarjee A, Ramful D, Randrianaivo H, Doray B, Faivre L, Amiel J, Campeau PM, Thevenon J. Recessive loss of function PIGN alleles, including an intragenic deletion with founder effect in La Reunion Island, in patients with Fryns syndrome. Eur J Hum Genet. 2018 Mar;26(3):340-349. doi: 10.1038/s41431-017-0087-x. Epub 2018 Jan 12. Citation on PubMed
- Cunniff C, Jones KL, Saal HM, Stern HJ. Fryns syndrome: an autosomal recessive disorder associated with craniofacial anomalies, diaphragmatic hernia, and distal digital hypoplasia. Pediatrics. 1990 Apr;85(4):499-504. Citation on PubMed
- Lin AE, Pober BR, Mullen MP, Slavotinek AM. Cardiovascular malformations in Fryns syndrome: is there a pathogenic role for neural crest cells? Am J Med Genet A. 2005 Dec 15;139(3):186-93. doi: 10.1002/ajmg.a.31023. Citation on PubMed
- Loong L, Tardivo A, Knaus A, Hashim M, Pagnamenta AT, Alt K, Bohrer-Rabel H, Caro-Llopis A, Cole T, Distelmaier F, Edery P, Ferreira CR, Jezela-Stanek A, Kerr B, Kluger G, Krawitz PM, Kuhn M, Lemke JR, Lesca G, Lynch SA, Martinez F, Maxton C, Mierzewska H, Monfort S, Nicolai J, Orellana C, Pal DK, Ploski R, Quarrell OW, Rosello M, Rydzanicz M, Sabir A, Smigiel R, Stegmann APA, Stewart H, Stumpel C, Szczepanik E, Tzschach A, Wolfe L, Taylor JC, Murakami Y, Kinoshita T, Bayat A, Kini U. Biallelic variants in PIGN cause Fryns syndrome, multiple congenital anomalies-hypotonia-seizures syndrome, and neurologic phenotypes: A genotype-phenotype correlation study. Genet Med. 2023 Jan;25(1):37-48. doi: 10.1016/j.gim.2022.09.007. Epub 2022 Nov 2. Citation on PubMed
- Moerman P, Fryns JP, Vandenberghe K, Devlieger H, Lauweryns JM. The syndrome of diaphragmatic hernia, abnormal face and distal limb anomalies (Fryns syndrome): report of two sibs with further delineation of this multiple congenital anomaly (MCA) syndrome. Am J Med Genet. 1988 Dec;31(4):805-14. doi: 10.1002/ajmg.1320310413. Citation on PubMed
- Neville HL, Jaksic T, Wilson JM, Lally PA, Hardin WD Jr, Hirschl RB, Langham MR Jr, Lally KP; Congenital Diaphragmatic Hernia Study Group. Fryns syndrome in children with congenital diaphragmatic hernia. J Pediatr Surg. 2002 Dec;37(12):1685-7. doi: 10.1053/jpsu.2002.36695. Citation on PubMed
- Ramsing M, Gillessen-Kaesbach G, Holzgreve W, Fritz B, Rehder H. Variability in the phenotypic expression of fryns syndrome: A report of two sibships. Am J Med Genet. 2000 Dec 18;95(5):415-24. doi: 10.1002/1096-8628(20001218)95:53.0.co;2-j. Citation on PubMed
- Slavotinek A, Lee SS, Davis R, Shrit A, Leppig KA, Rhim J, Jasnosz K, Albertson D, Pinkel D. Fryns syndrome phenotype caused by chromosome microdeletions at 15q26.2 and 8p23.1. J Med Genet. 2005 Sep;42(9):730-6. doi: 10.1136/jmg.2004.028787. Citation on PubMed or Free article on PubMed Central
- Slavotinek A. Fryns Syndrome. 2007 Apr 18 [updated 2025 Dec 4]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1459/ Citation on PubMed
- Slavotinek AM, Schauer G, Machin G, Dasouki M, Rueda-Pedraza ME, Chiricosta F, Keller R. Fryns syndrome: report of eight new cases. Genet Med. 2005 Jan;7(1):74-6. doi: 10.1097/01.gim.0000151337.68184.3f. No abstract available. Citation on PubMed
- Slavotinek AM. Fryns syndrome: a review of the phenotype and diagnostic guidelines. Am J Med Genet A. 2004 Feb 1;124A(4):427-33. doi: 10.1002/ajmg.a.20381. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.