

Description
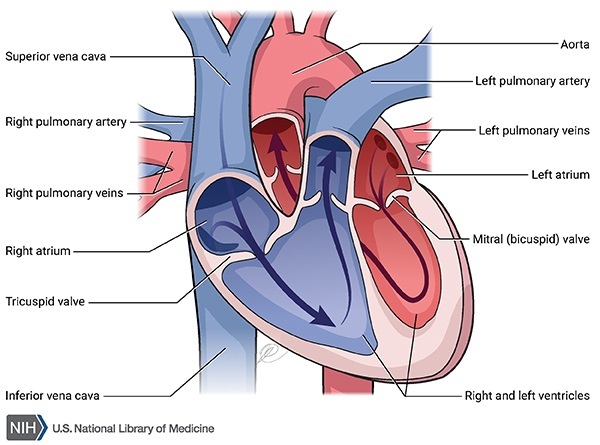
Familial restrictive cardiomyopathy is a genetic form of heart disease. For the heart to beat normally, the heart (cardiac) muscle must contract and relax in a coordinated way. Oxygen-rich blood from the lungs travels first through the upper chambers of the heart (the atria), and then to the lower chambers of the heart (the ventricles).
In people with familial restrictive cardiomyopathy, the heart muscle is stiff and cannot fully relax after each contraction. Impaired muscle relaxation causes blood to back up in the atria and lungs, which reduces the amount of blood in the ventricles.
Familial restrictive cardiomyopathy can appear anytime from childhood to adulthood. The first signs and symptoms of this condition in children are failure to gain weight and grow at the expected rate (failure to thrive), extreme tiredness (fatigue), and fainting. Children who are severely affected may also have abnormal swelling or puffiness (edema), increased blood pressure, an enlarged liver, an abnormal buildup of fluid in the abdominal cavity (ascites), and lung congestion. Some children with familial restrictive cardiomyopathy do not have any obvious signs or symptoms, but they may die suddenly due to heart failure. Without treatment, the majority of affected children survive only a few years after they are diagnosed.
Adults with familial restrictive cardiomyopathy typically first develop shortness of breath, fatigue, and a reduced ability to exercise. Some individuals have an irregular heart beat (arrhythmia) and may also experience a sensation of fluttering or pounding in the chest (palpitations) and dizziness. Abnormal blood clots are commonly seen in adults with this condition. Without treatment, approximately one-third of adults with familial restrictive cardiomyopathy do not survive more than five years after diagnosis.
Frequency
The prevalence of familial restrictive cardiomyopathy is unknown. Although cardiomyopathy is a relatively common condition, restrictive cardiomyopathy, in which relaxation of the heart muscle is impaired, is the least common type. Some other forms of cardiomyopathy involve a weak or enlarged heart muscle with impaired contraction. In the United States and in Europe, restrictive cardiomyopathy accounts for less than five percent of all cardiomyopathies. The proportion of restrictive cardiomyopathy that runs in families is not known.
Causes
Mutations in several genes have been found to cause familial restrictive cardiomyopathy. Mutations in the TNNI3 gene are one of the major causes of this condition. The TNNI3 gene provides instructions for making a protein called cardiac troponin I, which is found solely in the heart. Cardiac troponin I is one of three proteins that make up the troponin protein complex, which helps regulate tensing (contraction) and relaxation of the heart muscle.
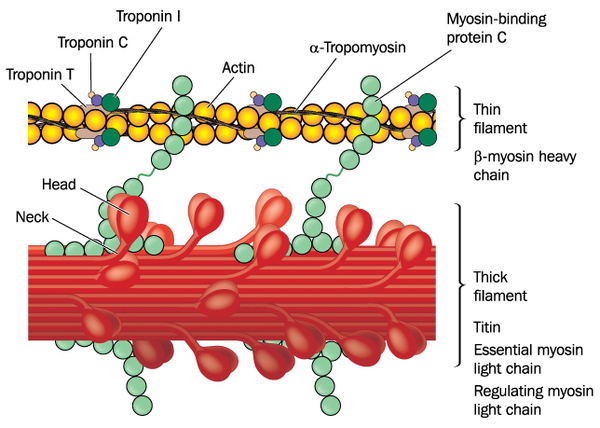
TNNI3 gene mutations associated with familial restrictive cardiomyopathy result in the production of a defective cardiac troponin I protein. The altered protein disrupts the function of the troponin protein complex and does not allow the heart muscle to fully relax. As a result, not enough blood enters the ventricles, leading to a buildup in the atria and lungs. The abnormal heart relaxation and blood flow is responsible for many of the signs and symptoms of familial restrictive cardiomyopathy.
Mutations in other genes associated with familial restrictive cardiomyopathy each account for a small percentage of cases of this condition. Some people with familial restrictive cardiomyopathy do not have an identified mutation in any of the known associated genes. The cause of the disorder in these individuals is unknown.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Cardiomyopathy, restrictive
- RCM
Additional Information & Resources
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008 Jan;29(2):270-6. doi: 10.1093/eurheartj/ehm342. Epub 2007 Oct 4. Citation on PubMed
- Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, Christiansen M, Andersen PS, Sebire N, Ashworth M, Deanfield JE, McKenna WJ, Elliott PM. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008 Nov;94(11):1478-84. doi: 10.1136/hrt.2007.134684. Epub 2008 May 8. Citation on PubMed
- Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009 May;24(3):214-20. doi: 10.1097/hco.0b013e32832a1d2e. No abstract available. Citation on PubMed
- Muchtar E, Blauwet LA, Gertz MA. Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res. 2017 Sep 15;121(7):819-837. doi: 10.1161/CIRCRESAHA.117.310982. Citation on PubMed
- Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009 Dec;10(8):iii23-33. doi: 10.1093/ejechocard/jep156. Citation on PubMed
- Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of restrictive cardiomyopathy. Heart Fail Clin. 2010 Apr;6(2):179-86. doi: 10.1016/j.hfc.2009.11.005. Citation on PubMed
- Xu Q, Dewey S, Nguyen S, Gomes AV. Malignant and benign mutations in familial cardiomyopathies: insights into mutations linked to complex cardiovascular phenotypes. J Mol Cell Cardiol. 2010 May;48(5):899-909. doi: 10.1016/j.yjmcc.2010.03.005. Epub 2010 Mar 16. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.