

Description
Familial hemophagocytic lymphohistiocytosis is a disorder in which the immune system produces too many activated immune cells (lymphocytes) called T cells, natural killer cells, B cells, and macrophages (histiocytes). Excessive amounts of immune system proteins called cytokines are also produced. This overactivation of the immune system causes fever and damages the liver and spleen, resulting in enlargement of these organs.
Familial hemophagocytic lymphohistiocytosis also destroys blood-producing cells in the bone marrow, a process called hemophagocytosis. As a result, affected individuals have low numbers of red blood cells (anemia) and a reduction in the number of platelets, which are involved in clotting. A reduction in platelets may cause easy bruising and abnormal bleeding.


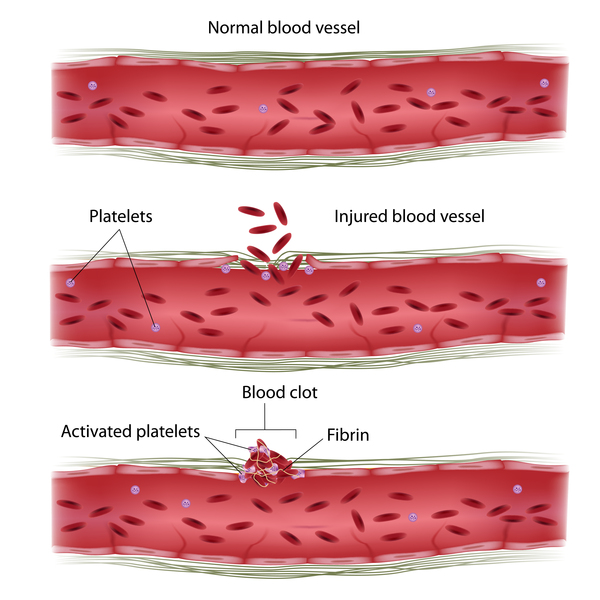
The brain may also be affected in familial hemophagocytic lymphohistiocytosis. As a result, affected individuals may experience irritability, delayed closure of the bones of the skull in infants, neck stiffness, abnormal muscle tone, impaired muscle coordination, paralysis, blindness, seizures, and coma. In addition to neurological problems, familial hemophagocytic lymphohistiocytosis can cause abnormalities of the heart, kidneys, and other organs and tissues. Affected individuals also have an increased risk of developing cancers of blood-forming cells (leukemia and lymphoma).
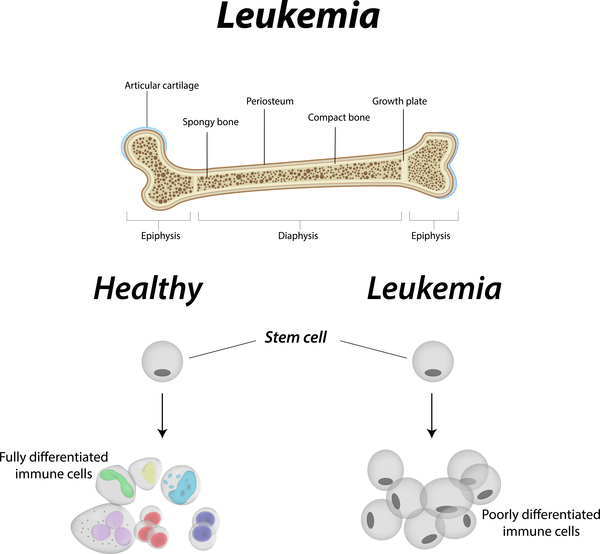
Signs and symptoms of familial hemophagocytic lymphohistiocytosis usually become apparent during infancy, although occasionally they appear later in life. They usually occur when the immune system launches an exaggerated response to an infection, but may also occur in the absence of infection. Without treatment, most people with familial hemophagocytic lymphohistiocytosis survive only a few months.
Frequency
Familial hemophagocytic lymphohistiocytosis occurs in approximately 1 in 50,000 individuals worldwide.
Causes
Familial hemophagocytic lymphohistiocytosis may be caused by mutations in any of several genes. These genes provide instructions for making proteins that help destroy or deactivate lymphocytes that are no longer needed. By controlling the number of activated lymphocytes, these genes help regulate immune system function.
Approximately 40 to 60 percent of cases of familial hemophagocytic lymphohistiocytosis are caused by mutations in the PRF1 or UNC13D genes. Smaller numbers of cases are caused by mutations in other known genes. In some affected individuals, the genetic cause of the disorder is unknown.
The gene mutations that cause familial hemophagocytic lymphohistiocytosis impair the body's ability to regulate the immune system. These changes result in the exaggerated immune response characteristic of this condition.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Familial erythrophagocytic lymphohistiocytosis
- Familial hemophagocytic histiocytosis
- Familial hemophagocytic lymphocytosis
- Familial hemophagocytic reticulosis
- FEL
- FHL
- FHLH
- Hemophagocytic syndrome
- HPLH
- Primary hemophagocytic hymphohistiocytosis
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS, FAMILIAL, 1; FHL1
- HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS, FAMILIAL, 4; FHL4
- HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS, FAMILIAL, 2; FHL2
- HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS, FAMILIAL, 3; FHL3
- HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS, FAMILIAL, 5, WITH OR WITHOUT MICROVILLUS INCLUSION DISEASE; FHL5
Scientific Articles on PubMed
References
- Cetica V, Pende D, Griffiths GM, Arico M. Molecular basis of familial hemophagocytic lymphohistiocytosis. Haematologica. 2010 Apr;95(4):538-41. doi: 10.3324/haematol.2009.019562. No abstract available. Citation on PubMed or Free article on PubMed Central
- Filipovich AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr Opin Allergy Clin Immunol. 2011 Dec;11(6):512-6. doi: 10.1097/ACI.0b013e32834c22f5. Citation on PubMed
- Horne A, Ramme KG, Rudd E, Zheng C, Wali Y, al-Lamki Z, Gurgey A, Yalman N, Nordenskjold M, Henter JI. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br J Haematol. 2008 Oct;143(1):75-83. doi: 10.1111/j.1365-2141.2008.07315.x. Epub 2008 Aug 15. Citation on PubMed
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233-46. doi: 10.1146/annurev-med-041610-134208. Citation on PubMed
- Marsh RA, Satake N, Biroschak J, Jacobs T, Johnson J, Jordan MB, Bleesing JJ, Filipovich AH, Zhang K. STX11 mutations and clinical phenotypes of familial hemophagocytic lymphohistiocytosis in North America. Pediatr Blood Cancer. 2010 Jul 15;55(1):134-40. doi: 10.1002/pbc.22499. Citation on PubMed
- Pagel J, Beutel K, Lehmberg K, Koch F, Maul-Pavicic A, Rohlfs AK, Al-Jefri A, Beier R, Bomme Ousager L, Ehlert K, Gross-Wieltsch U, Jorch N, Kremens B, Pekrun A, Sparber-Sauer M, Mejstrikova E, Wawer A, Ehl S, zur Stadt U, Janka G. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood. 2012 Jun 21;119(25):6016-24. doi: 10.1182/blood-2011-12-398958. Epub 2012 Mar 26. Citation on PubMed
- Santoro A, Cannella S, Trizzino A, Bruno G, De Fusco C, Notarangelo LD, Pende D, Griffiths GM, Arico M. Mutations affecting mRNA splicing are the most common molecular defect in patients with familial hemophagocytic lymphohistiocytosis type 3. Haematologica. 2008 Jul;93(7):1086-90. doi: 10.3324/haematol.12622. Epub 2008 May 19. Citation on PubMed
- Sieni E, Cetica V, Santoro A, Beutel K, Mastrodicasa E, Meeths M, Ciambotti B, Brugnolo F, zur Stadt U, Pende D, Moretta L, Griffiths GM, Henter JI, Janka G, Arico M. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis type 3. J Med Genet. 2011 May;48(5):343-52. doi: 10.1136/jmg.2010.085456. Epub 2011 Jan 19. Citation on PubMed or Free article on PubMed Central
- Trizzino A, zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, Beutel K, Sumegi J, Cannella S, Pende D, Mian A, Henter JI, Griffiths G, Santoro A, Filipovich A, Arico M; Histiocyte Society HLH Study group. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet. 2008 Jan;45(1):15-21. doi: 10.1136/jmg.2007.052670. Epub 2007 Sep 14. Citation on PubMed
- Zhang K, Astigarraga I, Bryceson Y, Lehmberg K, Machowicz R, Marsh R, Sieni E, Wang Z, Nichols KE. Familial Hemophagocytic Lymphohistiocytosis. 2006 Mar 22 [updated 2024 Jun 6]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1444/ Citation on PubMed
- Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, Villanueva J, Risma KA, Wei Q, Klein PS, Filipovich AH. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011 Nov 24;118(22):5794-8. doi: 10.1182/blood-2011-07-370148. Epub 2011 Aug 31. Citation on PubMed or Free article on PubMed Central
- Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, Hennies HC. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006 Jan;27(1):62-8. doi: 10.1002/humu.20274. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.