

Description
Factor XIII deficiency is a rare bleeding disorder. Researchers have identified an inherited form and a less severe form that is acquired during a person's lifetime.
Signs and symptoms of inherited factor XIII deficiency begin soon after birth, usually with abnormal bleeding from the umbilical cord stump. If the condition is not treated, affected individuals may have episodes of excessive and prolonged bleeding that can be life-threatening. Abnormal bleeding can occur after surgery or minor trauma. The condition can also cause spontaneous bleeding into the joints or muscles, leading to pain and disability. Women with inherited factor XIII deficiency tend to have heavy or prolonged menstrual bleeding (menorrhagia) and may experience recurrent pregnancy losses (miscarriages). Other signs and symptoms of inherited factor XIII deficiency include nosebleeds, bleeding of the gums, easy bruising, problems with wound healing, bleeding after surgery, and abnormal scar formation. Inherited factor XIII deficiency also increases the risk of spontaneous bleeding inside the skull (intracranial hemorrhage), which is the leading cause of death in people with this condition.
Acquired factor XIII deficiency becomes apparent later in life. People with the acquired form are less likely to have severe or life-threatening episodes of abnormal bleeding than those with the inherited form.
Frequency
Inherited factor XIII deficiency affects 1 to 3 per million people worldwide. Researchers suspect that mild factor XIII deficiency, including the acquired form of the disorder, is underdiagnosed because many affected people never have a major episode of abnormal bleeding that would lead to a diagnosis.
Causes
Inherited factor XIII deficiency results from mutations in the F13A1 gene or, less commonly, the F13B gene. These genes provide instructions for making the two parts (subunits) of a protein called factor XIII. This protein plays a critical role in the coagulation cascade, which is a series of chemical reactions that forms blood clots in response to injury. After an injury, clots seal off blood vessels to stop bleeding and trigger blood vessel repair. Factor XIII acts at the end of the cascade to strengthen and stabilize newly formed clots, preventing further blood loss.
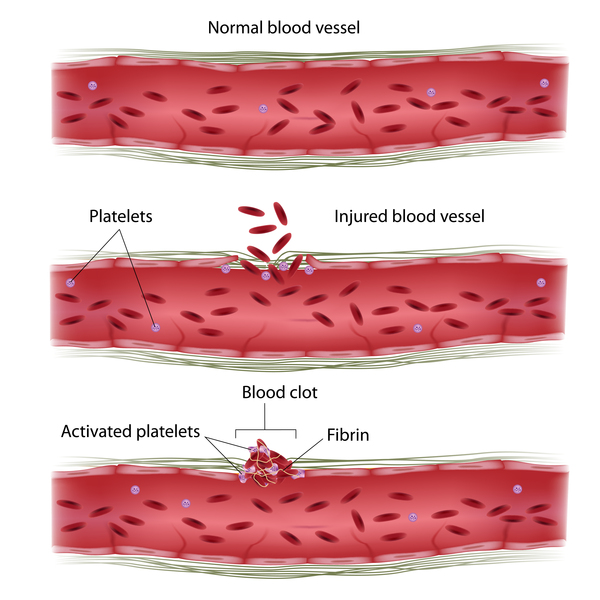
Mutations in the F13A1 or F13B gene significantly reduce the amount of functional factor XIII available to participate in blood clotting. In most people with the inherited form of the condition, factor XIII levels in the bloodstream are less than 5 percent of normal. A loss of this protein's activity weakens blood clots, preventing the clots from stopping blood loss effectively.
The acquired form of factor XIII deficiency results when the production of factor XIII is reduced or when the body uses factor XIII faster than cells can replace it. Acquired factor XIII deficiency is generally mild because levels of factor XIII in the bloodstream are 20 to 70 percent of normal; levels above 10 percent of normal are usually adequate to prevent spontaneous bleeding episodes.
Acquired factor XIII deficiency can be caused by disorders including an inflammatory disease of the liver called hepatitis, scarring of the liver (cirrhosis), inflammatory bowel disease, overwhelming bacterial infections (sepsis), and several types of cancer. Acquired factor XIII deficiency can also be caused by abnormal activation of the immune system, which produces specialized proteins called autoantibodies that attack and disable the factor XIII protein. The production of autoantibodies against factor XIII is sometimes associated with immune system diseases such as systemic lupus erythematosus and rheumatoid arthritis. In other cases, the trigger for autoantibody production is unknown.
Inheritance
Inherited factor XIII deficiency is considered to have an autosomal recessive pattern of inheritance, which means that it results when both copies of either the F13A1 gene or the F13B gene in each cell have mutations.

Some people, including parents of individuals with factor XIII deficiency, carry a single mutated copy of the F13A1 or F13B gene in each cell. These mutation carriers have a reduced amount of factor XIII in their bloodstream (20 to 60 percent of normal), and they may experience abnormal bleeding after surgery, dental work, or major trauma. However, most people who carry one mutated copy of the F13A1 or F13B gene do not have abnormal bleeding episodes under normal circumstances, and so they never come to medical attention.
The acquired form of factor XIII deficiency is not inherited and does not run in families.
Other Names for This Condition
- Deficiency of factor XIII
- Deficiency, Laki-Lorand factor
- Fibrin stabilizing factor deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Biswas A, Ivaskevicius V, Thomas A, Oldenburg J. Coagulation factor XIII deficiency. Diagnosis, prevalence and management of inherited and acquired forms. Hamostaseologie. 2014;34(2):160-6. doi: 10.5482/HAMO-13-08-0046. Epub 2014 Feb 7. Citation on PubMed
- de Jager T, Pericleous L, Kokot-Kierepa M, Naderi M, Karimi M. The burden and management of FXIII deficiency. Haemophilia. 2014 Nov;20(6):733-40. doi: 10.1111/hae.12474. Epub 2014 Jul 17. Citation on PubMed
- Kohler HP, Ichinose A, Seitz R, Ariens RA, Muszbek L; Factor XIII And Fibrinogen SSC Subcommittee Of The ISTH. Diagnosis and classification of factor XIII deficiencies. J Thromb Haemost. 2011 Jul;9(7):1404-6. doi: 10.1111/j.1538-7836.2011.04315.x. No abstract available. Citation on PubMed
- Levy JH, Greenberg C. Biology of Factor XIII and clinical manifestations of Factor XIII deficiency. Transfusion. 2013 May;53(5):1120-31. doi: 10.1111/j.1537-2995.2012.03865.x. Epub 2012 Aug 28. Citation on PubMed
- Muszbek L, Bagoly Z, Cairo A, Peyvandi F. Novel aspects of factor XIII deficiency. Curr Opin Hematol. 2011 Sep;18(5):366-72. doi: 10.1097/MOH.0b013e3283497e3e. Citation on PubMed
- Schroeder V, Kohler HP. Factor XIII deficiency: an update. Semin Thromb Hemost. 2013 Sep;39(6):632-41. doi: 10.1055/s-0033-1353392. Epub 2013 Aug 8. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.