

Description
Early-onset myopathy with fatal cardiomyopathy (EOMFC) is an inherited muscle disease that affects the skeletal muscles, which are used for movement, and the heart (cardiac) muscle. This condition is characterized by skeletal muscle weakness that becomes apparent in early infancy. Affected individuals have delayed development of motor skills, such as sitting, standing, and walking. Beginning later in childhood, people with EOMFC may also develop joint deformities called contractures that restrict the movement of the neck and back. Scoliosis, which is an abnormal side-to-side curvature of the spine, also develops in late childhood.
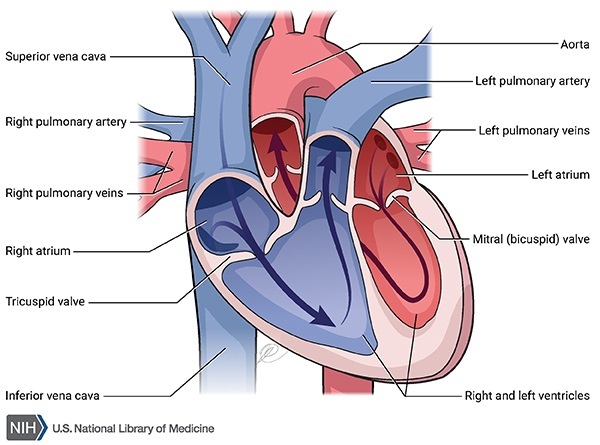

A form of heart disease called dilated cardiomyopathy is another feature of EOMFC. Dilated cardiomyopathy enlarges and weakens the cardiac muscle, preventing the heart from pumping blood efficiently. Signs and symptoms of this condition can include an irregular heartbeat (arrhythmia), shortness of breath, extreme tiredness (fatigue), and swelling of the legs and feet. The heart abnormalities associated with EOMFC usually become apparent in childhood, after the skeletal muscle abnormalities. The heart disease worsens quickly, and it often causes heart failure and sudden death in adolescence or early adulthood.
Frequency
EOMFC appears to be a rare disorder, although its prevalence is unknown. It has been reported in a small number of families of Moroccan and Sudanese descent.
Causes
EOMFC is caused by variants (also known as mutations) in the TTN gene. This gene provides instructions for making a protein called titin, which plays an important role in skeletal and cardiac muscle function.
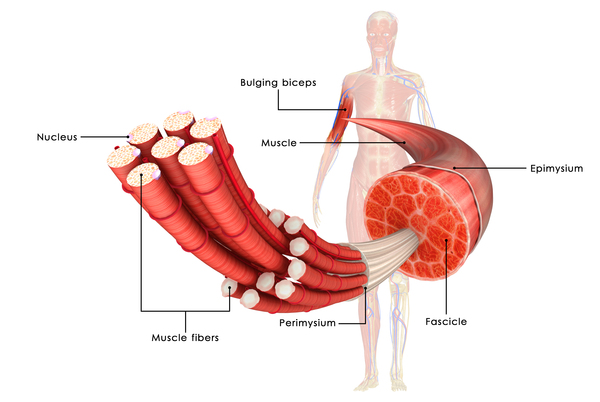
Within muscle cells, titin is an essential component of structures called sarcomeres and is critical for muscles to tense (contract) normally . Sarcomeres are the basic units of muscle contraction; they are made of proteins that generate the mechanical force needed for muscles to contract. Titin has several functions within sarcomeres. One of this protein's most important jobs is to provide structure, flexibility, and stability to these cell structures. Titin also plays a role in chemical signaling and in assembling new sarcomeres.
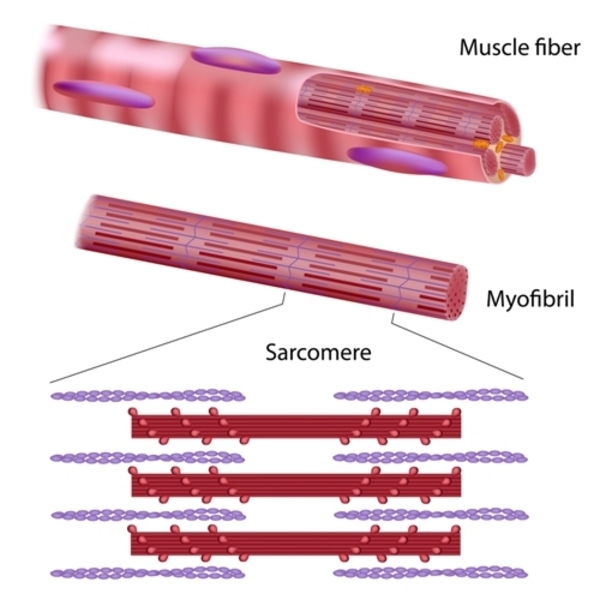
The TTN gene variants involved in EOMFC lead to the production of an abnormally short version of titin. The abnormal protein disrupts the function of sarcomeres, which prevents skeletal and cardiac muscle from contracting normally. These muscle abnormalities underlie the features of EOMFC, including skeletal muscle weakness and dilated cardiomyopathy.

Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- EOMFC
- Salih CMD
- Salih congenital muscular dystrophy
- Salih myopathy
- Titinopathy & early-onset myopathy with fatal cardiomyopathy
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Carmignac V, Salih MA, Quijano-Roy S, Marchand S, Al Rayess MM, Mukhtar MM, Urtizberea JA, Labeit S, Guicheney P, Leturcq F, Gautel M, Fardeau M, Campbell KP, Richard I, Estournet B, Ferreiro A. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol. 2007 Apr;61(4):340-51. doi: 10.1002/ana.21089. Citation on PubMed
- Hackman P, Savarese M, Di Feo MF, Udd B, Salih MA. Salih Myopathy. 2012 Jan 12 [updated 2025 Apr 3]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK83297/ Citation on PubMed
- Salih MA, Al Rayess M, Cutshall S, Urtizberea JA, Al-Turaiki MH, Ozo CO, Straub V, Akbar M, Abid M, Andeejani A, Campbell KP. A novel form of familial congenital muscular dystrophy in two adolescents. Neuropediatrics. 1998 Dec;29(6):289-93. doi: 10.1055/s-2007-973579. Citation on PubMed
- Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P. Increasing Role of Titin Mutations in Neuromuscular Disorders. J Neuromuscul Dis. 2016 Aug 30;3(3):293-308. doi: 10.3233/JND-160158. Citation on PubMed or Free article on PubMed Central
- Subahi SA. Distinguishing cardiac features of a novel form of congenital muscular dystrophy (Salih cmd). Pediatr Cardiol. 2001 Jul-Aug;22(4):297-301. doi: 10.1007/s002460010233. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.