

Description
Dystrophic epidermolysis bullosa is one of the major forms of a group of conditions called epidermolysis bullosa. Epidermolysis bullosa cause the skin to be very fragile and to blister easily. Blisters and skin erosions form in response to minor injury or friction, such as rubbing or scratching. The signs and symptoms of dystrophic epidermolysis bullosa vary widely among affected individuals. In mild cases, blistering may primarily affect the hands, feet, knees, and elbows. Severe cases of this condition involve widespread blistering that can lead to vision loss, scarring, and other serious medical problems.
Researchers classify dystrophic epidermolysis bullosa into major types based on the inheritance pattern and features of the condition. Although the types differ in severity, their features overlap significantly and they are caused by mutations in the same gene.
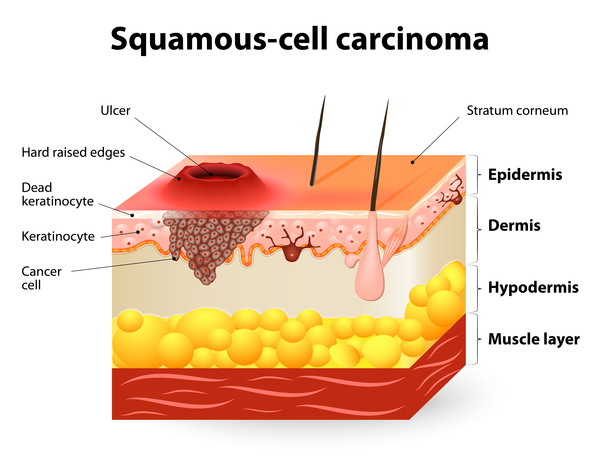
Recessive dystrophic epidermolysis bullosa severe generalized (RDEB-sev gen) is the classic form of the condition and is the most severe. Affected infants are typically born with widespread blistering and areas of missing skin, often caused by trauma that occurs during birth. Most often, blisters are present over the whole body and affect mucous membranes such as the moist lining of the mouth and digestive tract. As the blisters heal, they result in severe scarring. Scarring in the mouth and esophagus can make it difficult to chew and swallow food, leading to chronic malnutrition and slow growth. Additional complications of ongoing scarring can include fusion of the skin between the fingers and toes, loss of fingernails and toenails, joint deformities (contractures) that restrict movement, and eye inflammation leading to vision loss. Additionally, people with RDEB-sev gen have a very high risk of developing a form of skin cancer called squamous cell carcinoma in young adulthood. In these individuals, the cancer tends to be unusually aggressive and is often life-threatening.
Other types of recessive dystrophic epidermolysis bullosa fall along a spectrum referred to as RDEB-generalized and localized (RDEB-gen and -loc). These forms of the condition are somewhat less severe than RDEB-sev gen and are distinguished by the affected regions of the body. Blistering is often limited to the hands, feet, knees, and elbows in mild cases, but may be widespread in more severe cases. Rare forms affect specific regions of the body, such as the shins or the abdomen. Affected people often have malformed fingernails and toenails. The RDEB-gen and -loc types involve scarring in the areas where blisters occur, but these forms of the condition do not cause the severe scarring characteristic of RDEB-sev gen.
Another major type of this condition is known as dominant dystrophic epidermolysis bullosa (DDEB). The signs and symptoms of this condition tend to be milder than those of the recessive forms, with blistering often limited to the hands, feet, knees, and elbows. The blisters heal with scarring, but it is less severe than in recessive forms of this condition. Most affected people have malformed fingernails and toenails, and the nails may be lost over time. In the mildest cases, abnormal nails are the only sign of the condition.
Frequency
Considered together, the prevalence of recessive and dominant dystrophic epidermolysis bullosa is estimated to be 3.3 per million people.
Causes
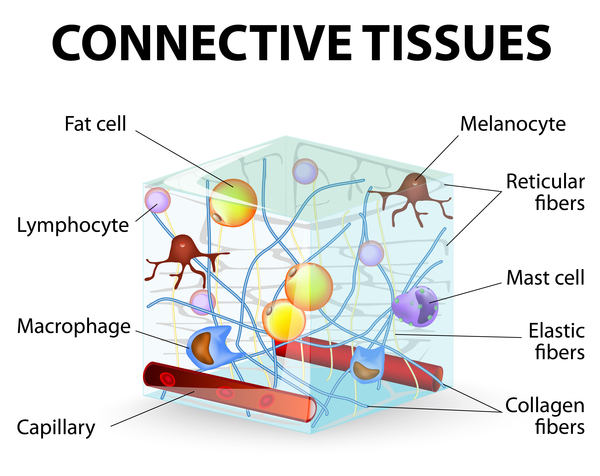
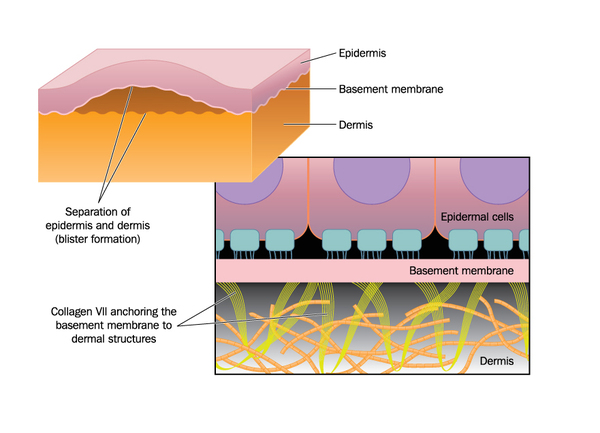
Mutations in the COL7A1 gene cause all forms of dystrophic epidermolysis bullosa. This gene provides instructions for making a protein that forms the pieces (subunits) of a larger protein called type VII collagen. Collagens are proteins that give structure and strength to connective tissues, such as skin, tendons, and ligaments, throughout the body. Type VII collagen plays an important role in strengthening and stabilizing the skin. It is the main component of structures called anchoring fibrils, which anchor the top layer of skin, called the epidermis, to an underlying layer called the dermis.
COL7A1 gene mutations alter the structure or disrupt the production of the type VII collagen subunit protein. These changes affect the production of type VII collagen. Mutations that severely reduce or prevent the production of type VII collagen cause RDEB-sev gen. Mutations that allow a small amount of normal or partially functional type VII collagen to be produced lead to milder forms of the dystrophic epidermolysis bullosa. When type VII collagen is abnormal or missing, the formation of anchoring fibrils is impaired. A shortage of these fibrils disrupts the connection of the epidermis to the dermis, and friction or other minor trauma can cause the two skin layers to separate. This separation leads to the formation of blisters, which can cause extensive scarring as they heal. Researchers are working to determine how abnormalities of type VII collagen also underlie the increased risk of skin cancer seen in RDEB-sev gen.
Inheritance
Recessive dystrophic epidermolysis bullosa (RDEB-sev gen and RDEB-gen and -loc) is inherited in an autosomal recessive pattern. Autosomal recessive inheritance means that both copies of the COL7A1 gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.

DDEB has an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder. About 70 percent of all people with DDEB have inherited an altered COL7A1 gene from an affected parent. The remaining 30 percent of affected people have the condition as a result of a new mutation in the COL7A1 gene. These cases occur in people with no history of the disorder in their family.


Other Names for This Condition
- DEB
- Epidermolysis bullosa dystrophica
- Epidermolysis bullosa, dystrophic
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Das BB, Sahoo S. Dystrophic epidermolysis bullosa. J Perinatol. 2004 Jan;24(1):41-7. doi: 10.1038/sj.jp.7211019. No abstract available. Citation on PubMed
- De Benedittis M, Petruzzi M, Favia G, Serpico R. Oro-dental manifestations in Hallopeau-Siemens-type recessive dystrophic epidermolysis bullosa. Clin Exp Dermatol. 2004 Mar;29(2):128-32. doi: 10.1111/j.1365-2230.2004.01485.x. Citation on PubMed
- Dharma B, Moss C, McGrath JA, Mellerio JE, Ilchyshyn A. Dominant dystrophic epidermolysis bullosa presenting as familial nail dystrophy. Clin Exp Dermatol. 2001 Jan;26(1):93-6. Citation on PubMed
- Dystrophic Epidermolysis Bullosa Research Association of America (DebRA)
- Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, Marinkovich MP, Martinez AE, McGrath JA, Mellerio JE, Moss C, Murrell DF, Shimizu H, Uitto J, Woodley D, Zambruno G. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014 Jun;70(6):1103-26. doi: 10.1016/j.jaad.2014.01.903. Epub 2014 Mar 29. Citation on PubMed
- Fine JD. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016 Nov 1;152(11):1231-1238. doi: 10.1001/jamadermatol.2016.2473. Citation on PubMed
- Lucky AW, Pope E, Crawford S. Dystrophic Epidermolysis Bullosa. 2006 Aug 21 [updated 2025 Aug 7]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1304/ Citation on PubMed
- Mallipeddi R, Bleck O, Mellerio JE, Ashton GH, Eady RA, McGrath JA. Dilemmas in distinguishing between dominant and recessive forms of dystrophic epidermolysis bullosa. Br J Dermatol. 2003 Oct;149(4):810-8. doi: 10.1046/j.1365-2133.2003.05315.x. Citation on PubMed
- Soro L, Bartus C, Purcell S. Recessive dystrophic epidermolysis bullosa: a review of disease pathogenesis and update on future therapies. J Clin Aesthet Dermatol. 2015 May;8(5):41-6. Citation on PubMed or Free article on PubMed Central
- Uitto J, Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetic classification. Clin Dermatol. 2005 Jan-Feb;23(1):33-40. doi: 10.1016/j.clindermatol.2004.09.015. Citation on PubMed
- Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet. 2007 Mar;44(3):181-92. doi: 10.1136/jmg.2006.045302. Epub 2006 Sep 13. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.