

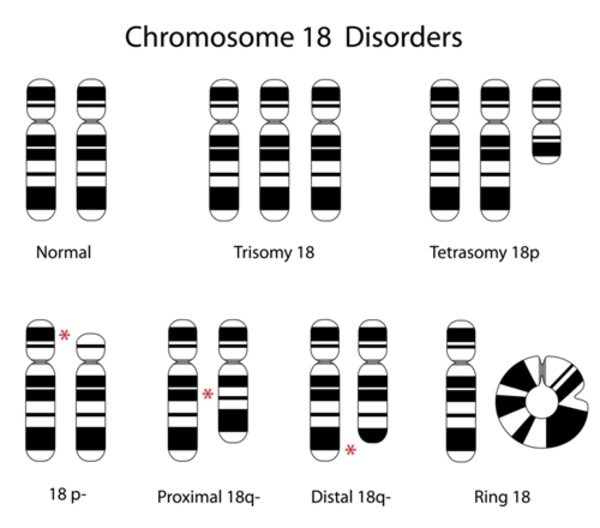
Description
Distal 18q deletion syndrome is a chromosomal condition that occurs when a piece of the long (q) arm of chromosome 18 is missing. The term "distal" means that the missing piece occurs near one end of the chromosome. Distal 18q deletion syndrome can lead to a wide variety of signs and symptoms among affected individuals.
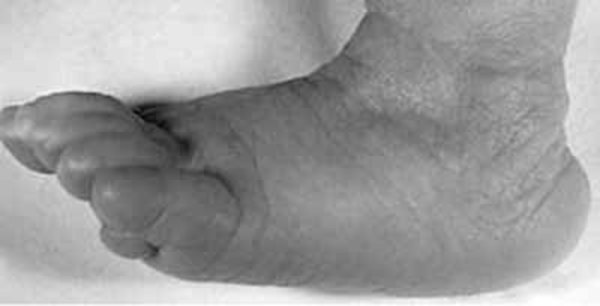
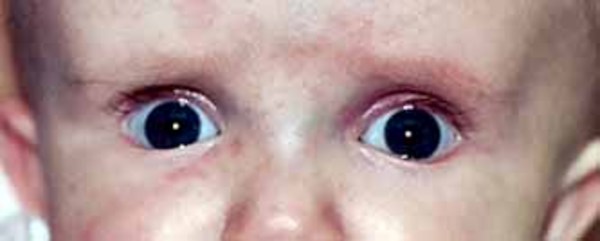
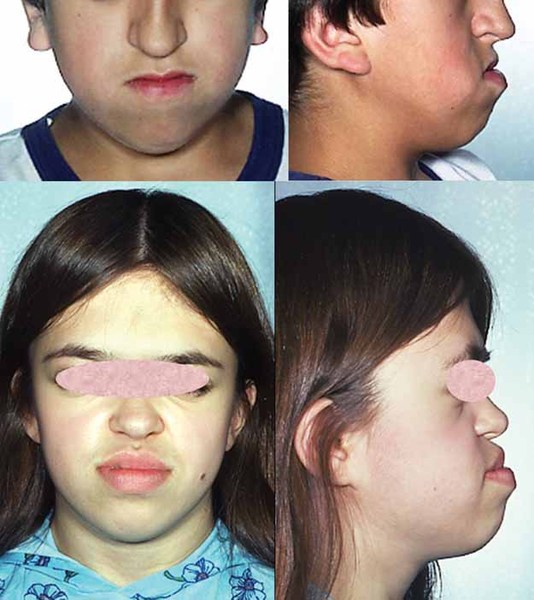
Some common features of distal 18q deletion syndrome include short stature (often due to growth hormone deficiency), weak muscle tone (hypotonia), hearing loss due to ear canals that are narrow (aural stenosis) or absent (aural atresia), and foot abnormalities such as an inward or upward-turning foot (clubfoot) or feet with soles that are rounded outward (rocker-bottom feet). Eye movement disorders and other vision problems, an opening in the roof of the mouth (cleft palate), an underactive thyroid gland (hypothyroidism), heart abnormalities that are present from birth (congenital heart defects), kidney problems, genital abnormalities, and skin problems may also occur in this disorder. Some affected individuals have mild facial differences such as deep-set eyes, a flat or sunken appearance of the middle of the face (midface hypoplasia), a wide mouth, and prominent ears. These features are often not noticeable except in a detailed medical evaluation.


Distal 18q deletion syndrome can also affect the nervous system. A common neurological feature of this disorder is impaired myelin production (dysmyelination). Myelin is a fatty substance that insulates nerve cells and promotes the rapid transmission of nerve impulses. The formation of a protective myelin sheath around nerve cells (myelination) normally begins before birth and continues into adulthood. In people with distal 18q deletion syndrome, myelination is often delayed and proceeds more slowly than normal; affected individuals may never have normal adult myelin levels. Most people with distal 18q deletion syndrome have neurological problems, although it is unclear to what extent these problems are related to the dysmyelination. These problems include delayed development, learning disabilities, and intellectual disability that can range from mild to severe. Seizures; hyperactivity; mood disorders such as anxiety, irritability, and depression; and features of autism spectrum disorder that affect communication and social interaction may also occur. Some affected individuals have an unusually small head size (microcephaly).

Frequency
Deletions from the q arm of chromosome 18 occur in an estimated 1 in 55,000 newborns worldwide. Most of these deletions occur in the distal region of the q arm, leading to distal 18q deletion syndrome.
Causes
Distal 18q deletion syndrome is caused by a deletion of genetic material from one copy of chromosome 18 anywhere between a region called 18q21 and the end of the chromosome. The size of the deletion and where it begins vary among affected individuals. The signs and symptoms of distal 18q deletion syndrome are thought to be related to the loss of multiple genes, some of which have not been identified, from this part of chromosome 18. Certain features of the disorder have been associated with the loss of particular genes in this region. People with deletions that include the TCF4 gene usually have signs and symptoms of another genetic condition known as Pitt-Hopkins syndrome, such as severe intellectual disability and breathing problems, in addition to other features of distal 18q deletion syndrome.
Inheritance
Distal 18q deletion syndrome is considered to be an autosomal dominant condition, which means one copy of the deleted region on chromosome 18 in each cell is sufficient to cause the disorder's characteristic features.
Most cases of distal 18q deletion syndrome are the result of a new (de novo) deletion and are not inherited. The deletion occurs most often as a random event during the formation of reproductive cells (eggs or sperm) or in early fetal development. Affected people typically have no history of the disorder in their family.

In some cases, distal 18q deletion syndrome is inherited, usually from an affected parent with relatively mild signs and symptoms. The condition can also be inherited from an unaffected parent who carries a chromosomal rearrangement called a balanced translocation, in which no genetic material is gained or lost. Individuals with a balanced translocation do not usually have any related health problems; however, the translocation can become unbalanced as it is passed to the next generation. Children who inherit an unbalanced translocation can have a chromosomal rearrangement with extra or missing genetic material. Inheritance of an unbalanced translocation that results in the deletion of genetic material from the distal region of the q arm of chromosome 18 causes distal 18q deletion syndrome.
Other Names for This Condition
- 18q deletion syndrome
- 18q- syndrome
- Chromosome 18 long arm deletion syndrome
- Chromosome 18q deletion syndrome
- Chromosome 18q monosomy
- Chromosome 18q- syndrome
- Del(18q) syndrome
- Monosomy 18q
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Cody JD, Sebold C, Heard P, Carter E, Soileau B, Hasi-Zogaj M, Hill A, Rupert D, Perry B, O'Donnell L, Gelfond J, Lancaster J, Fox PT, Hale DE. Consequences of chromsome18q deletions. Am J Med Genet C Semin Med Genet. 2015 Sep;169(3):265-80. doi: 10.1002/ajmg.c.31446. Epub 2015 Aug 3. Citation on PubMed
- Daviss WB, O'Donnell L, Soileau BT, Heard P, Carter E, Pliszka SR, Gelfond JA, Hale DE, Cody JD. Mood disorders in individuals with distal 18q deletions. Am J Med Genet B Neuropsychiatr Genet. 2013 Dec;162B(8):879-88. doi: 10.1002/ajmg.b.32197. Epub 2013 Sep 4. Citation on PubMed
- Feenstra I, Vissers LE, Orsel M, van Kessel AG, Brunner HG, Veltman JA, van Ravenswaaij-Arts CM. Genotype-phenotype mapping of chromosome 18q deletions by high-resolution array CGH: an update of the phenotypic map. Am J Med Genet A. 2007 Aug 15;143A(16):1858-67. doi: 10.1002/ajmg.a.31850. Citation on PubMed
- Hale DE, Cody JD, Baillargeon J, Schaub R, Danney MM, Leach RJ. The spectrum of growth abnormalities in children with 18q deletions. J Clin Endocrinol Metab. 2000 Dec;85(12):4450-4. doi: 10.1210/jcem.85.12.7016. Citation on PubMed
- Hasi M, Soileau B, Sebold C, Hill A, Hale DE, O'Donnell L, Cody JD. The role of the TCF4 gene in the phenotype of individuals with 18q segmental deletions. Hum Genet. 2011 Dec;130(6):777-87. doi: 10.1007/s00439-011-1020-y. Epub 2011 Jun 14. Citation on PubMed or Free article on PubMed Central
- Lancaster JL, Cody JD, Andrews T, Hardies LJ, Hale DE, Fox PT. Myelination in children with partial deletions of chromosome 18q. AJNR Am J Neuroradiol. 2005 Mar;26(3):447-54. Citation on PubMed
- Linnankivi T, Tienari P, Somer M, Kahkonen M, Lonnqvist T, Valanne L, Pihko H. 18q deletions: clinical, molecular, and brain MRI findings of 14 individuals. Am J Med Genet A. 2006 Feb 15;140(4):331-9. doi: 10.1002/ajmg.a.31072. Citation on PubMed
- Linnankivi TT, Autti TH, Pihko SH, Somer MS, Tienari PJ, Wirtavuori KO, Valanne LK. 18q-syndrome: brain MRI shows poor differentiation of gray and white matter on T2-weighted images. J Magn Reson Imaging. 2003 Oct;18(4):414-9. doi: 10.1002/jmri.10383. Citation on PubMed
- Nuijten I, Admiraal R, Van Buggenhout G, Cremers C, Frijns JP, Smeets D, van Ravenswaaij-Arts C. Congenital aural atresia in 18q deletion or de Grouchy syndrome. Otol Neurotol. 2003 Nov;24(6):900-6. doi: 10.1097/00129492-200311000-00014. Citation on PubMed
- Schaub RL, Hale DE, Rose SR, Leach RJ, Cody JD. The spectrum of thyroid abnormalities in individuals with 18q deletions. J Clin Endocrinol Metab. 2005 Apr;90(4):2259-63. doi: 10.1210/jc.2004-1630. Epub 2005 Jan 25. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.