

Description
Dilated cardiomyopathy with ataxia (DCMA) syndrome is an inherited condition characterized by heart problems, movement difficulties, and other features affecting multiple body systems.
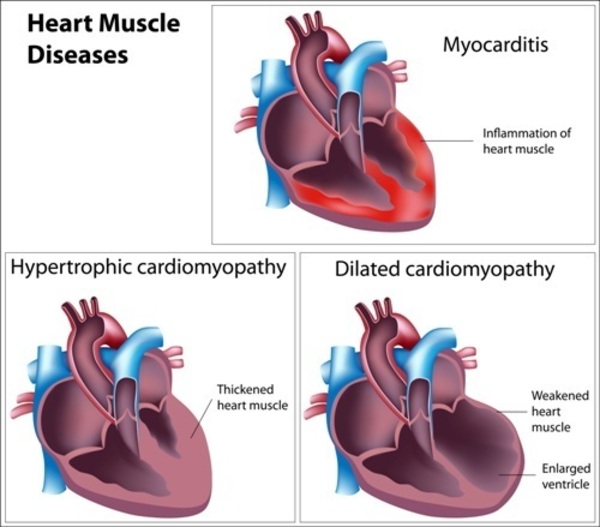
Beginning in infancy to early childhood, most people with DCMA syndrome develop dilated cardiomyopathy, which is a condition that weakens and enlarges the heart, preventing it from pumping blood efficiently. Some affected individuals also have long QT syndrome, which is a heart condition that causes the cardiac muscle to take longer than usual to recharge between beats. The irregular heartbeats (arrhythmia) can lead to fainting (syncope) or cardiac arrest and sudden death. Rarely, heart problems improve over time; however, in most cases of DCMA syndrome, affected individuals do not survive past childhood due to heart failure. A small percentage of people with DCMA syndrome have no heart problems at all.
By age 2, children with DCMA syndrome have problems with coordination and balance (ataxia). These movement problems can result in delay of motor skills such as standing and walking, but most older children with DCMA syndrome can walk without support.
In addition to heart problems and movement difficulties, most individuals with DCMA syndrome grow slowly before and after birth, which leads to short stature. Additionally, many affected individuals have mild intellectual disability. Many males with DCMA syndrome have genital abnormalities such as undescended testes (cryptorchidism) or the urethra opening on the underside of the penis (hypospadias). Other common features of DCMA syndrome include unusually small red blood cells (microcytic anemia), which can cause pale skin; an abnormal buildup of fats in the liver (hepatic steatosis), which can damage the liver; and the degeneration of nerve cells that carry visual information from the eyes to the brain (optic nerve atrophy), which can lead to vision loss.


DCMA syndrome is associated with increased levels of a substance called 3-methylglutaconic acid in the urine. The amount of acid does not appear to influence the signs and symptoms of the condition. DCMA syndrome is one of a group of metabolic disorders that can be diagnosed by the presence of increased levels of 3-methylglutaconic acid in urine (3-methylglutaconic aciduria). People with DCMA syndrome also have high urine levels of another acid called 3-methylglutaric acid.
Frequency
DCMA syndrome is a very rare disorder. Approximately 30 cases have been identified in the Dariusleut Hutterite population of the Great Plains region of Canada. Only a few affected individuals have been identified outside this population.
Causes
Mutations in the DNAJC19 gene cause DCMA syndrome. The DNAJC19 gene provides instructions for making a protein found in structures called mitochondria, which are the energy-producing centers of cells. While the exact function of the DNAJC19 protein is unclear, it may regulate the transport of other proteins into and out of mitochondria.
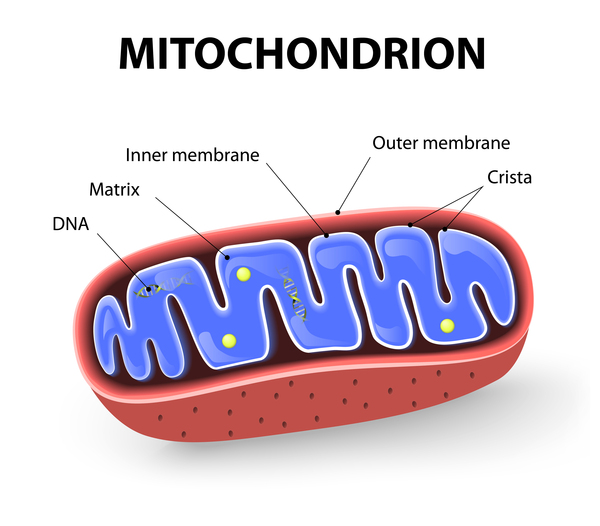
The DNAJC19 gene mutations that cause DCMA syndrome lead to the production of an abnormally shortened protein that likely has impaired function. Researchers speculate that a lack of functional DNAJC19 protein alters the transport of other proteins into and out of the mitochondria. When too many or too few proteins move in and out of the mitochondria, energy production and mitochondrial survival can be reduced. Tissues that have high energy demands, such as the heart and the brain, are especially susceptible to decreases in cellular energy production. It is likely that this loss of cellular energy damages these and other tissues, leading to heart problems, movement difficulties, and other features of DCMA syndrome.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- 3-methylglutaconic aciduria type V
- DCMA
- DCMA syndrome
- DNAJC19 defect
- MGA type V
- MGA5
- MGCA5
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, Snyder FF, Bridge PJ, Bernier FP. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet. 2006 May;43(5):385-93. doi: 10.1136/jmg.2005.036657. Epub 2005 Jul 31. Citation on PubMed or Free article on PubMed Central
- Ojala T, Polinati P, Manninen T, Hiippala A, Rajantie J, Karikoski R, Suomalainen A, Tyni T. New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatr Res. 2012 Oct;72(4):432-7. doi: 10.1038/pr.2012.92. Epub 2012 Jul 13. Citation on PubMed
- Sparkes R, Patton D, Bernier F. Cardiac features of a novel autosomal recessive dilated cardiomyopathic syndrome due to defective importation of mitochondrial protein. Cardiol Young. 2007 Apr;17(2):215-7. doi: 10.1017/S1047951107000042. Epub 2007 Jan 23. Citation on PubMed
- Wortmann SB, Duran M, Anikster Y, Barth PG, Sperl W, Zschocke J, Morava E, Wevers RA. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis. 2013 Nov;36(6):923-8. doi: 10.1007/s10545-012-9580-0. Epub 2013 Jan 8. Citation on PubMed
- Wortmann SB, Kluijtmans LA, Engelke UF, Wevers RA, Morava E. The 3-methylglutaconic acidurias: what's new? J Inherit Metab Dis. 2012 Jan;35(1):13-22. doi: 10.1007/s10545-010-9210-7. Epub 2010 Sep 30. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.