

Description
Cystinosis is a condition characterized by accumulation of the amino acid cystine (a building block of proteins) within cells. Excess cystine damages cells and often forms crystals that can build up and cause problems in many organs and tissues. The kidneys and eyes are especially vulnerable to damage; the muscles, thyroid, pancreas, and testes may also be affected.
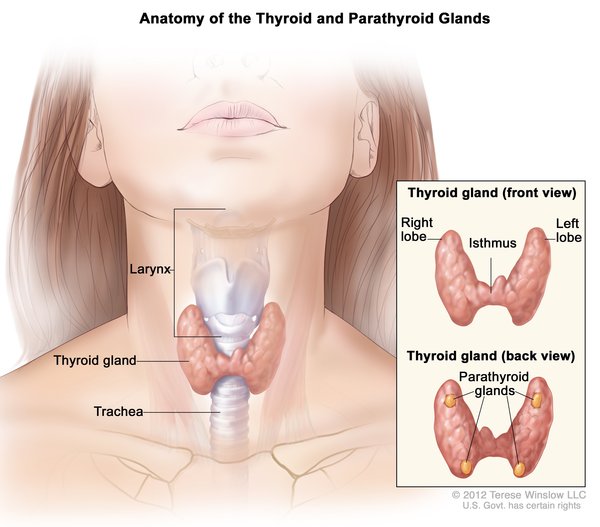
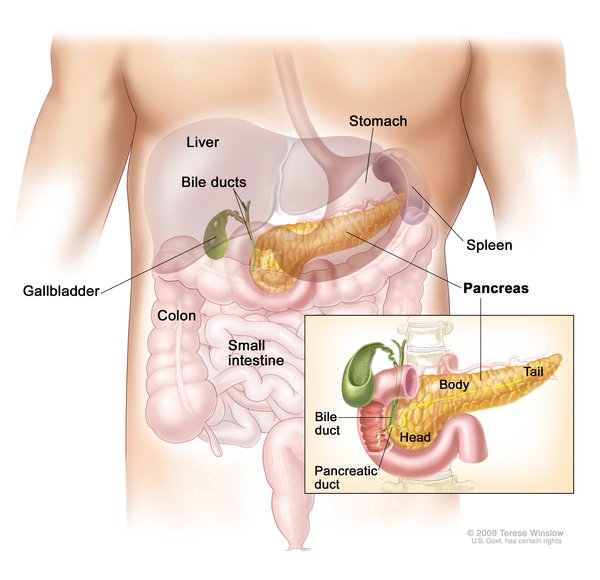
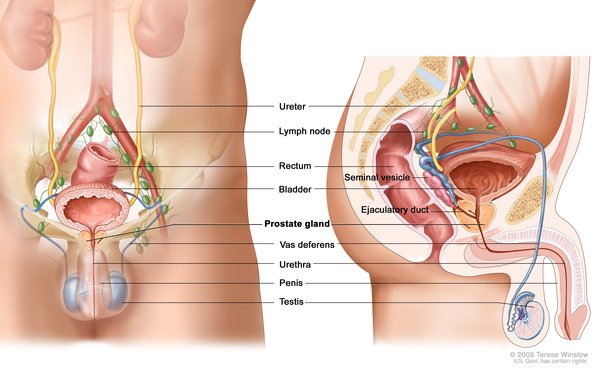
There are three distinct types of cystinosis. In order of decreasing severity, they are nephropathic cystinosis, intermediate cystinosis, and non-nephropathic or ocular cystinosis.
Nephropathic cystinosis begins in infancy, causing poor growth and a particular type of kidney damage (renal Fanconi syndrome) in which certain molecules that should be reabsorbed into the bloodstream are instead eliminated in the urine. The kidney problems lead to the loss of important minerals, salts, fluids, and many other nutrients. The loss of nutrients impairs growth and may result in soft, bowed bones (hypophosphatemic rickets), especially in the legs. The nutrient imbalances in the body lead to increased urination, thirst, dehydration, and abnormally acidic blood (acidosis). By about the age of 2, cystine crystals may be present in the clear covering of the eye (cornea). The buildup of these crystals in the eye causes pain and an increased sensitivity to light (photophobia). Untreated children will experience complete kidney failure by about the age of 10. Other signs and symptoms that may occur in untreated people, especially after adolescence, include muscle deterioration, blindness, inability to swallow, diabetes, thyroid and nervous system problems, and an inability to father children (infertility) in affected men.

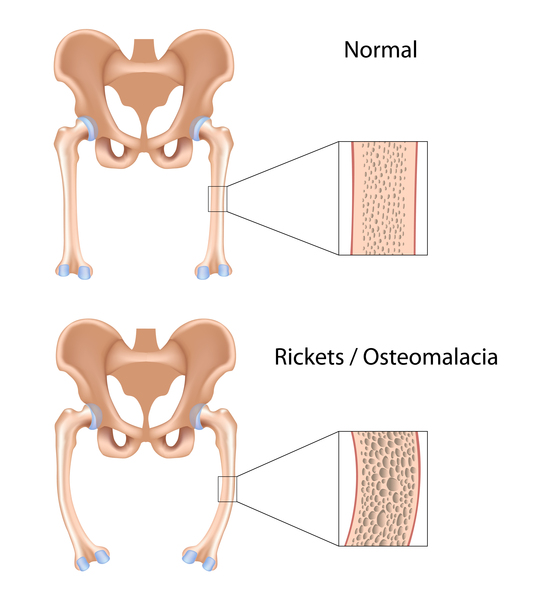
The signs and symptoms of intermediate cystinosis are the same as nephropathic cystinosis, but they occur at a later age. Intermediate cystinosis typically becomes apparent in affected individuals in adolescence. Malfunctioning kidneys and corneal crystals are the main initial features of this disorder. If intermediate cystinosis is left untreated, complete kidney failure will occur, but usually not until the late teens to mid-twenties.
People with non-nephropathic or ocular cystinosis typically experience photophobia due to cystine crystals in the cornea, but usually do not develop kidney malfunction or most of the other signs and symptoms of cystinosis. Due to the absence of severe symptoms, the age at which this form of cystinosis is diagnosed varies widely.
Frequency
Cystinosis affects approximately 1 in 100,000 to 200,000 newborns worldwide. The incidence is higher in the province of Brittany, France, where the disorder affects 1 in 26,000 individuals.
Causes
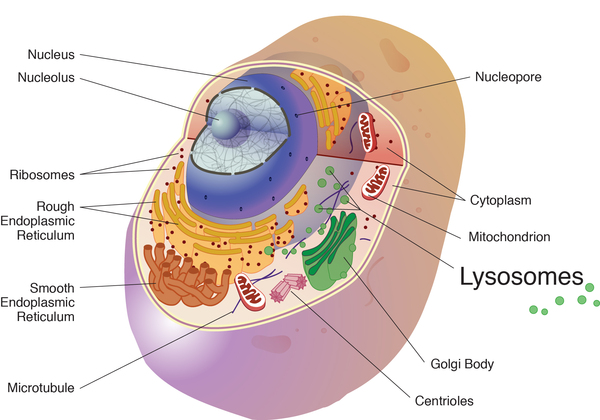
All three types of cystinosis are caused by mutations in the CTNS gene. Mutations in this gene lead to a deficiency of a transporter protein called cystinosin. Within cells, this protein normally moves cystine out of the lysosomes, which are compartments in the cell that digest and recycle materials. When cystinosin is defective or missing, cystine accumulates and forms crystals in the lysosomes. The buildup of cystine damages cells in the kidneys and eyes and may also affect other organs.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Cystine storage disease
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Delgado G, Schatz A, Nichols S, Appelbaum M, Trauner D. Behavioral profiles of children with infantile nephropathic cystinosis. Dev Med Child Neurol. 2005 Jun;47(6):403-7. doi: 10.1017/s0012162205000782. Citation on PubMed
- Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002 Jul 11;347(2):111-21. doi: 10.1056/NEJMra020552. No abstract available. Citation on PubMed
- Kalatzis V, Antignac C. New aspects of the pathogenesis of cystinosis. Pediatr Nephrol. 2003 Mar;18(3):207-15. doi: 10.1007/s00467-003-1077-5. Epub 2003 Feb 27. Citation on PubMed
- Kleta R, Gahl WA. Cystinosis: antibodies and healthy bodies. J Am Soc Nephrol. 2002 Aug;13(8):2189-91. doi: 10.1097/01.asn.0000027098.90648.f2. No abstract available. Citation on PubMed
- Kleta R, Gahl WA. Pharmacological treatment of nephropathic cystinosis with cysteamine. Expert Opin Pharmacother. 2004 Nov;5(11):2255-62. doi: 10.1517/14656566.5.11.2255. Citation on PubMed
- Kleta R, Kaskel F, Dohil R, Goodyer P, Guay-Woodford LM, Harms E, Ingelfinger JR, Koch VH, Langman CB, Leonard MB, Mannon RB, Sarwal M, Schneider JA, Skovby F, Sonies BC, Thoene JG, Trauner DA, Gahl WA; NIH Office of Rare Diseases. First NIH/Office of Rare Diseases Conference on Cystinosis: past, present, and future. Pediatr Nephrol. 2005 Apr;20(4):452-4. doi: 10.1007/s00467-004-1777-5. Epub 2005 Jan 27. No abstract available. Citation on PubMed
- Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol. 2008 Jun;23(6):863-78. doi: 10.1007/s00467-007-0650-8. Citation on PubMed
- Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol. 2013 Jan;28(1):51-9. doi: 10.1007/s00467-012-2242-5. Epub 2012 Aug 18. Citation on PubMed or Free article on PubMed Central
- Servais A, Moriniere V, Grunfeld JP, Noel LH, Goujon JM, Chadefaux-Vekemans B, Antignac C. Late-onset nephropathic cystinosis: clinical presentation, outcome, and genotyping. Clin J Am Soc Nephrol. 2008 Jan;3(1):27-35. doi: 10.2215/CJN.01740407. Citation on PubMed or Free article on PubMed Central
- Sonies BC, Almajid P, Kleta R, Bernardini I, Gahl WA. Swallowing dysfunction in 101 patients with nephropathic cystinosis: benefit of long-term cysteamine therapy. Medicine (Baltimore). 2005 May;84(3):137-146. doi: 10.1097/01.md.0000164204.00159.d4. Citation on PubMed
- Tsilou E, Zhou M, Gahl W, Sieving PC, Chan CC. Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis: report of a case and review of the literature. Surv Ophthalmol. 2007 Jan-Feb;52(1):97-105. doi: 10.1016/j.survophthal.2006.10.006. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.